前言
免疫细胞化学(Immunocytochemistry)又称免疫组织化学(Immunohistochemistry)是组织化学的分支,它是用标记的特异性抗体(或抗原)对组织内抗原(或抗体)的分布进行细胞和组织原位的检测技术。Coons及其同事们于1941年首次用荧光素标记抗体检测肺组织内肺炎双球菌获得成功,开创了细胞化学中“免疫细胞化学”这一新篇章 。免疫细胞化学的迅猛发展是在近10余年。继Nakan建立的酶标记抗体技术后, Sternberger在此基础上改良并建立了辣根过氧化物酶-抗过氧化物酶(PAP)技术,使免疫细胞化学得到日益广泛的应用。80年代,Hsu等建立了抗生物素-生物素(ABC)法之后,免疫金-银染色法、半抗原标记法、免疫电镜技术等相继问世,使免疫细胞化学技术成为当今生物医学中形态、功能、代谢综合研究的一项有力工具,近年来,随着抗原的提纯和抗体标记技术的改进,特别是单克隆抗体技术的引入,使免疫细胞化学在生物基础研究如病理学、神经科学、发育生物学、细胞生物学和微生物、寄生虫、病毒等病原体的诊断和研究中日益显示出巨大的实用价值,并使实验向临床,向定量和分子水平深入。鉴于免疫细胞化学的发展极为迅速,我们在原编写的“实用免疫细胞化学”一书的基础上汇入了免疫细胞化学国内外最新进展并增编核酸分子杂交技术。希望本书能有益于我国生命科学的研究,并能给读者新的信息和启示。
本书以理论与开发技术相结合,实用为主 ,尽力兼顾技术方法的先进性和科学性,全书正文廿三章 及附录1~4,计约84万字,105幅插图。第一章 为总论,简述免疫细胞化学的发展概况、有关基础理论和技术。第二章 叙述了抗原和抗体的制备。第三至八章 分别详述了免疫荧光细胞化学、免疫酶细胞化学、免疫金、银和铁标记细胞化学、亲和免疫细胞化学。电子显微镜免疫细胞化学和蛋白质及多肽激素的放射免疫测定步骤、原位投射自显影技术等第十一至十七章 分别介绍免疫细胞化学在神经科学、肿瘤病学、消化器官、皮肤病学、自身免疫性疾病和病原体的快速检查方法及应用。
近10多年来,相继发现了多种亲合物质,如植物凝集素(lectins)与糖结合物(glycol conju-gates)、葡萄球菌A蛋白(staphylococal protein A)与IgG、生物素( biotin)与卵白素(亲合素)(A-vidin)、激素、脂质与受体等。这些物质是一些有多价结合能力的物质,不但亲和物质之间有高度亲合力,而且可以与标记物如荧光素、酶、同位素、铁蛋白等相结合。Bayer(1976)将这种利用两种物质之间的高度亲合力而相互结合的化学反应,进行细胞化学检测,称为亲合细胞化学(affinity cytochemistry)。它和免疫细胞化学的区别是亲合物质之间的结合不是抗原和抗体反应。然而抗原和抗体反应也是一种物质间的亲合反应,是一种特殊的亲合细胞化学。由于引入亲合细胞化学,提高和增加了免疫细胞化学的敏感性和检测范围,是免疫细胞化学的新发展,本书第六章 予详述。
原位分子杂交(In situ hybridization)技术引入免疫细胞化学,是现代免疫细胞化学向基因水平深入发展的重要标志。近年来,分子杂交与免疫细胞化学的结合日益紧密,尤其是在杂交后信号的检测技术中,引入了免疫细胞化学的免疫放大和显色技术,已经形成了杂交免疫细胞化学或称原位杂交免疫细胞化学(In situ hybridizationimmunocytochemistry),成为免疫细胞化学中又一新的分支。分子杂交与免疫细胞化学的结合,形成了在细胞、亚细胞和分子水平同时检测基因及其表达产物的完整体系,把基因探针(基因工程)和单克隆抗体技术结合起来,成为当代生物科学和医学在分子水平研究和诊断的新兴技术。尤其是近年来与PCR技术结合的原位PCR技术和原位免疫PCR技术敏感性很高。本书第十八至二十三章 对有关分子生物学基础理论、核酸分子探针制备、原位分子杂交技术、聚合酶链式反应(PCR)、核酸分子杂交电镜技术、杂交与免疫细胞化学双标记技术、分子杂交的FISH技术和PRINS技术及其在染色体制片的应用和分子杂交基因诊断的应用等方面,由有关专家分别作出了专章 叙述。
定量免疫细胞化学技术是这个领域发展很快的一个方面,由于流式细胞仪,激光共聚焦显微仪和图像分析仪在免疫细胞化学中的应用,使免疫细胞化学的定量检测成为可能,从而提高了免疫细胞化学的定量水平。本书第九和十章 作了详细叙述。
本书附录列有一些常用的试剂配制方法,以便实际应用时参考。
由于本书编者都具有实际操作和应用免疫细胞化学与核酸分子杂交技术的经验,对某些方法的操作技术多结合编者的经验和在国内条件下应用的体会加以叙述,实用性强,能指导操作和启发读者如何应用。本书既有基础理论又有本领域国内外最新进展,对生物科学,尤其对医学科学工作者具有重要的参考价值,又可作为研究生、进修生的教材。
本书编写过程中,得到第三、四军医大学领导及广大同行的鼓励和支持;第三军医大学学报编辑室及张吉强、孙榆等同志在协助本书的出版、校对和抄写等方面做了大量的工作,在此深表感谢。
本书中难免有不当多处,恳切希望读者批评指正。
第一章 总论
免疫细胞化学又称免疫组织化学、其主要原理是用标记的抗体(或抗原)对细胞或组织内的相应抗原(或抗体)进行定性、定位或定量检测,经过组织化学的呈色反应之后,用显微镜、荧光显微镜或电子显微镜观察。凡是能作抗原、半抗原的物质,如蛋白质、多肽、核酸、酶、激素、磷脂、多糖、受体及病原体等都可用相应的特异性抗体的组织、细胞内将其用免疫细胞化学手段检出和研究。
免疫细胞化学的突出优点是:
1.高度特异性 抗原抗体反应是特异性最强的反应之一,免疫细胞化学所用的抗体必须是特异性强的多价或单价抗体,具有高度的识别能力,在抗原识别上可达到单个氨基酸的水平,这是其它组织化学难以相比的。
2.敏感性高 现代免疫细胞化学采用各种有效方法最大限度保存细胞和组织内待检物质的抗原性,或采用各种增敏方法,使用高度敏感的和高亲和力的抗体,保证可检出细胞内超微量的抗原成分,用显微镜或电子显微镜观察结果,因此,它是在细胞、分子水平或基因水平的检测技术。
3.方法步骤统一 若掌握一种技术操作方法步骤,则一通百通。
4.形态、机能和代谢密切结合 是一种综合定性、定位和定量密切结合;形态、机能和代谢密切结合为一体的研究和检测技术。
免疫细胞化学技术在细胞、染色体或亚细胞水平原位检测抗原分子,是其它任何生物技术难以达到和代替的,它以在细胞、基因和分子水平同时原位显示基因及其表达产物,形成了新的检测系统,为生物学、医学和各个领域分子水平的研究与诊断,开拓了广阔的前景。
免疫细胞化学技术日新月异,以惊人的速度发展。尤其是和分子生物学的理论和技术日益结合密切,基因探针、核酸分子杂交技术、原位PCR技术、核酸杂交和免疫细胞化学双标记技术、电镜杂交技术等成为免疫细胞化学技术的新发展成员,标志着免疫细胞化学又进入一个新的发展阶段。核酸分子探针-杂交-免疫细胞化学放大和显示杂交信号,可以称为杂交免疫细胞化学。所以,核酸分子探针的研制已成为免疫细胞化学实验室中必需的试剂生产技术,需要学习引进分子生物学的有关技术和设备,建立相应的基因工程实验条件,把基因重组技术、单克隆抗体技术和免疫细胞化学技术融合在一起,就形成了现代免疫细胞化学的新主体。更高倍电镜技术和图像分析,流式细胞仪技术和激光共聚焦显微技术和不断更新,把原位定性、定位和定量技术提高到了更新的水平,形成了免疫细胞化学发展的两翼,向着生命科学更广阔领域飞翔。
免疫细胞化学的全过程包括:①抗原提取和纯化;②免疫动物或细胞融合(单克隆抗体);③抗体效价检测和提取;④标记抗体;⑤细胞和组织切片标志的制备;⑥免疫细胞化学反应和显色;⑦观察和记录结果。
由此可见,免疫细化学的基本理论是抗原抗体反应,标记化学反应和呈色化学反应。由于免疫细胞化学和原位核酸分子杂交是在细胞和组织上进行抗原抗体反应,所以,必须熟练掌握显微标本制备的全过程,要求待检标本形态结构和抗原性保存良好,抗原不从原位扩散或丢失。所以从事免疫细胞化学的工作者必须了解以下有关理论及掌握有关细胞和组织学技术。
第一节 有关的免疫学理论(缺)
第二节 免疫细胞化学的有关技术概述
根据标记物的不同,免疫细胞化学技术可分为免疫荧光细胞化学技术,免疫酶细胞化学技术,免疫铁蛋白技术,免疫金-银细胞化学技术,亲和免疫细胞化学技术,免疫电子显微镜技术等。近些年来,核酸分子原位杂交技术采用生物素、地高辛等非放射性物质标记探针,和免疫细胞化学技术密切结合,发展为杂交免疫细胞化学技术。不同的免疫细胞化学技术,各具有独特的试剂和方法,但其基本技术方法是相似的,都包括抗休的制备,组织材料的处理、免疫染色、对照试验、显微镜观察等步骤。还有双重和多重标记技术也有重要的用途。
一、抗体的制备和配制
(一)抗体的制备
这是免疫细胞化学技术的首要试剂,必需制备具有很高特异性和敏感性的高效价抗体,这将在本书第二章 中详述。目前国内外市场各种特异性抗体日益增多,许多实验室直接应用市售产品,现在我国自制抗体种类少,发展抗体生产十分必要。尤其要定位一种新的抗原物质,能够自制较好。
(二)抗体的配制
包括抗体贮存液和抗体使用液的配制方法。新制备或购进的是原液或冻干品,抗血清为全血清,单克隆抗体是培养上清液或腹水。
1.抗体贮存获得新抗体后,应先根据生产厂家提供的抗体效价,将其分装,可每10μl或100μl/支分装入安瓿或0.25ml带盖塑料管中,密封。放入-20。C~40。C冰箱中保存备用,一般可保存1~2年。小量分装的抗体可1次用完,避免反复冻融而影响效价的降低。一般用前新鲜配制使用液体,稀释的抗体不能长时间保存,在4。C可存入1~3天,超过7天效价显著降低。
2.抗体使用液的配制这是任何免疫细胞化学方法中最重要的一环,无论是一抗、二抗和各种标记抗体,用前都必须按不同免疫染色方法和抗原性强弱与抗原的多少,稀释使用的各种抗体原液,以便获得最佳免疫染色结果。
(1)抗体最佳稀释度的测定方法:用已知阳性抗原切片,进行免疫染色,将其阳性强度与背景染色强度以“+”表示,可分为++++、+++、++、+、(-)。++++为最强阳性,+++为强阳性,++为较强阳性,+为弱阳性,(-)为阴性。
①直接测定法:用于测定第一抗体的最佳稀释度,其它条件稳定可靠。将一抗稀释为1:50、1:100、1:2001:400、1:500等5个稀释度滴加在阳性抗原切片上,同时设一替代和阴性对照,结果如表1-1:
表 1-1 选择最佳稀释抗血清方法
| 一抗稀释度 | 特异性染色强度 | 非特异性背景染色度 |
| 1:50 | ++++ | ++ |
| 1:100 | ++++ | ++ |
| 1:200 | ++++ | ++ |
| 1:400 | +++ | + |
| 1:500 | ++ | (-) |
| 阴性对照 | (-) | (-) |
从表中结果可见,第一抗体稀释到1:400时阳性结果呈强阳性,背景染色减少,其最佳稀释度在1:400~500之间。再作1:420、1:440、1:460、1:480、1:500稀释后染色,找出最佳稀释度。
②棋盘(方阵)测定方法:当测定两种以上抗体的最佳配合稀释度时,必须采用此法(见表1-2)。
表1-2 两种以上抗体最佳配合稀释度选择表
| 第二抗体 | 第 一 抗 体 | |||
| 1:500 | 1:1000 | 1:2000 | 1:4000 | |
| 1:100 | ++++(++) | ++++(+) | +++(±) | +(-) |
| 1:200 | ++++(+) | +++(-) | ++(-) | ++(-) |
| 1:400 | ++(-) | +(-) | - | - |
括号内为背景染色结果
从表中可见第一抗体1:1000,第二抗体1:2000接近最佳稀释度,再将一 抗体作1:600、1:700、1:800、1:900和1:1000稀释,即可找出最佳稀释度。
(2)抗体稀释液的配制:常用0.01mol/l pH7.4PBS或TBS缓冲液作抗体稀释液。可用以下方法配制专用的抗体稀释液,防止抗体效价下降,减少抗体在组织上的非特异性吸附:取0.05mol/l pH7.4 TBS100ml,加温到60。C,再加入优质明胶100mg,搅拌溶解后,冷却至室温,加入1g牛血清白蛋白,加入NaN3200mg溶解后,过滤,分装,4。C保存。
抗体的最佳稀释度由于各种抗体的效价不同,组织中抗原强弱不一,应根据不同情况作适当的调整,以取得中等阳性稀释度为佳,因其既适合于抗原性强和含量多的标本,也可用于抗原性弱的标本。
二、组织材料的处理
组织材料的处理是获得良好免疫组织化学结果的前提,必需保证要检测的细胞或组织取材新鲜,固定及时,形态保存完好,抗原物质的抗原性不丢失、不扩散和被破坏(下节 详述)。
三、免疫染色
可在细胞涂片或组织切片上进行免疫染色。一般程序是:①标记抗体与标本中抗原反应结合;②用PBS洗去未结合的成分;③直接观察结果(免疫荧光直接法);或显色后再用显微镜观察(免疫酶直接法)。在此基础上发展出间接法,多层法,双标记法等各种方法,将在本书各有关章 节 内详述。
在免疫染色中应特别注意增强特异性染色,减少或消除非特异性染色。在各种免疫染色中都必须注意以下几个问题:
1.增强特异性染色的方法
(1)蛋白酶消化法:其作用是暴露抗原,增加细胞和组织的通透性,以便抗体与抗原最大限度的结合,增强特异性染色和避免非特异性染色。这种方法已广泛用于各种免疫细胞化学染色,常用的蛋白酶有胰蛋白酶、胃蛋白酶以及链霉蛋白酶(pronase)等;也可用3mol/L尿素处理切片,达到酶消化的目的。各种酶的配制和使用方法详见附录。酶消化的时间和温度因各种抗原对消化的敏感性不同,应根据酶的活性通过预试验确定,消化的时间还与组织固定的时间有关,一般是陈旧固定组织所需时间长,以37。C为宜。消化时间短的组织可在室温中进行。消化处理时间过长能损伤组织,易使切片脱落,应使用切片粘附剂,消化时间尽量缩短。
(2)合适的抗体稀释度:抗体的浓度是免疫染色的关键,如果抗体浓度过高,抗体分子过多于抗原决定簇,可导致抗体结合减少,产生阴性结果。此阴性结果并不一定缺少抗原,而是由于抗体过量。这种现象类似于凝集反应中的前带效应(Prozone effect )。因此,必须使用一系列稀释作“棋盘式效价滴定”检测抗体的合适稀释度,以得到最大强度的特异性染色和最弱的背景染色。抗体稀释度应根据:①抗体效价高,溶液中特异性抗体浓度越高,工作稀释度越高;②一般讲,应用的抗体稀释度越大,温育时间越长。③抗体中非特异性蛋白含量、只有高稀释度时才能防止非特异性背景染色;④稀释用缓冲液的种类、标本的固定和处理过程等也可影响稀释度。所以合适的稀释度应根据自己的情况测定。抗体的稀释主要是指第一抗体,因为第一抗体中特异性抗体合适的尝试是关键,应用高稀释度第一抗体仅显示主亲和力的特异性染色反应,减少或消除其中交叉抗体反应。
(3)温育时间:大部分抗体温育时间为30-60min,必要时可4。C过夜(约18h)。温育的温度常用37。C,也可在室温中进行,对抗原抗体反应强的以室温为佳。37。C可增强抗原抗体反应;适用于多数抗体染色,但应注意在湿盒中进行,防止切片干燥而导致失败。
(4)多层染色法:对弱的抗原可用间接法(双层)、PAP和ABC法(三层)、四或五层PAP法或ABC法,或PAP和ABC联合染色法等,可以很大程度的提高敏感性,获得良好结果。
(5)显色增敏剂的应用,如在过氧化物酶底物中加入氯化镍,可提高显色敏感度4倍。
2.减少或消除非特异性染色的方法组织中非抗原抗体反应出现的阳性染色称为非特异性背景染色,最常见的原因是蛋白吸附于高电荷的胶元和结缔组织成分上。最有效方法是在用第一抗体前加制备第二抗体动物之非免疫血清(1:5-1:20)封闭组织上带电荷基团而除去与第一抗体非特异性结合。必要时可加入2%-5%牛血清白蛋白,可进一步减少非特异性染色。作用时间为10-20min。也可用除制备第一抗体以外的其它动物血清(非免疫的)。有明显溶血的血清不能用,以免产生非特异性染色。免疫荧光染色时,可用0.01%伊文氏兰(PBS溶液)稀释荧光抗体,对消除背景的非特异性荧光染色有很好的效果。当然使用特异性高、效价高的第一抗体是最重要的条件。洗涤用的缓冲液中加入0.85%~1%NaCl成为高盐溶液,充分洗涤切片,能有效的减少非特异性结合而减少背景染色。
3.显色反应的控制免疫酶染色应注意控制:①成色质浓度和温育时间可调节 ,增加成色质的量和/或增加底物温育时间,可增加反应产物强度。着色太深可减少温育反应时间。②过氧化物酶显色时,H2O2较大浓度将使显色反应过快而致背景加深;过量H2O2可能抑制酶的活性。
4.复染根据所用的染色方法和呈显颜色等,可选用适当的复染方法。如阳性结果呈红或棕色,则用苏木素将细胞核染成兰色,以便定位检测。也可用1%~2%甲基绿复染。
四、对照
其目的在于证明和肯定阳性结果的特异性,排除非特异性疑问。主要是针对第一抗体对照,常用的对照方法包括:①阳性对照;②阴性对照;③阻断试验;④替代对照;⑤空白对照;⑥自身对照;⑦吸收试验。
(一)阳性对照
用已知抗原阳性的切片与待检标本同时进行免疫细胞化学染色,对照切片应呈阳性结果,称为阳性对照。证明全过程均符合要求,尤其当待检标本呈阴性结果时,阳性对照尤为重要。
(二)阴性对照
用确证不含已知抗原的标本作对照,应呈阴性结果,称阴性对照,是阴性对照的一种。其实空白、替代、吸收和抑制试验都属阴性对照。当待检标本呈阳性结果时,阴性对照就更加重要,用以排除假阳性。对照的具体方法步骤,在有关章 节 详述。
五、免疫细胞化学结果的判断
对免疫细胞化学结果的判断应持科学的慎重态度,要准确判断阳性和阴性,排除假阳性和假阴性结果,必须严格对照实验,对新发现的阳性结果,除有对照试验结果之外,应进行多次重复实验,要求用几种方法进行验证,如用PAP法阳性,可再用ABC法验证。必须学会判断特异性染色和非特异性染色,对初学者更为重要,否则会得出不科学的结论。特异性染色与非特异性染色的鉴别点主要在于特异性反应产物常分布于特定的部位,如胞浆内,也有分布在细胞核和细胞表面的,即具有结构性。特异性染色表现为在同一切片上呈现不同程度的阳性染色结果。非特异性染色表现为无一定的分布规律,常为某一部位成片的均匀着色,细胞和周围的结缔组织均无区别的着色,或结缔组织呈现很强的染色。非特异性染色常出现在干燥切片的边缘,有刀痕或组织折叠的部位。在过大的组织块,中心固定不良也会导致非特异性染色。有时可见非特异性染色和特异性染色同时存在,由于过强的非特异性染色背景不但影响对特异性染色结果的观察和记录,而且令人对其特异性结果产生怀疑。
(一)阳性细胞的染色特征
免疫细胞化学的呈色深浅可反映抗原存在的数量,可作为定性、定位和定量的依据。
(1)阳性细胞染色分布有三种类型:①胞浆;②细胞核;③细胞膜表面。大部分抗原见于细胞浆,可见于整个胞浆或部分胞浆。
(2)阳性细胞分布可分为烟性和弥漫性。
(3)由于细胞内含抗原量的不同,所以染色强度不一。如果细胞之间染色强度相同,常提示其反应为非特异性。
(4)阳性细胞染色定位于细胞,且与阴性细胞相互交杂分布;而非特异性染色常不限于单个细胞,而是累及一片细胞。
(5)切片边缘、刀痕或皱折区域,坏死或挤压的细胞区,胶原结缔组织等,常表现为相同的阳性染色强度,不能用于判断阳性。
(二)染色失败的几种原因
(1)所染的全部切片均为阴性结果:包括阳性对照在内,全部呈阴性反应,原因可能是:①染色未严格按操作步骤进行;②漏加一种抗体,或抗体失效;③缓冲液内含叠氮化钠,抑制了酶的活性;④底物中所加H2O2量少或失效;⑤复染或脱水剂使用不当。
(2)所有切片均呈弱阳性反应:①切片在染色过程中抗体过浓,或干燥了;②缓冲液配制中未加氯化钠和pH值不准确,洗涤不彻底;③使用已变色的呈色底物溶液,或呈色反应时间过长;④抗体温育的时间过长;⑤H2O2浓度过高,呈色速度过快;⑥粘附剂太厚。
(3)所有切片背景过深;①未用酶消化处理切片;②切片或涂片过厚;③漂洗不够;④底物呈色反应过久;⑤蛋白质封闭不够或所用血清溶血;⑥使用全血清抗体稀释不够。
(4)阳性对照染色良好,检测的阳性标本呈阴性反应,固定和处理不当是最常见的原因。
对于阳性结果的定量判断常规方法是根据呈色深浅和阳性细胞数量分类计数,以(-)、+、++、+++等分级和计数统计。现在已采用图象分析计量,本书第九章 将详细叙述这种使免疫细胞化学的定量成为可能的先进的形态定量方法。另外,第十章 将详细途述另一种先进的免疫细胞化学计量分类术-流式细胞光度计技术。
第三节 有关细胞和组织学技术
一、细胞和组织
免疫组织化学技术是用标记物或显色物标记的抗体检测细胞和组织内的抗原,从而达到诊断和研究疾病的目的。抗原的准确显示和定位与制备的细胞和组织标本质量的好坏有着密切的联系。由于各种抗原的生化、物理性质不同,如温度高低、酸碱度强弱及各种化学试剂的作用均可影响抗原的免疫学活性,良好的细胞和组织学结构将有助于抗原的准确定位。因此,细胞和组织标本的采集制备在免疫组织化学技术中占有十分重要的位置。
(一)细胞标本的取材
目前,免疫组化技术已经应用于细胞学诊断,如鉴别低分化癌与恶性淋巴瘤、黑色素瘤、低分化肉瘤等。CEA应用于胸腹水中间皮瘤与癌的鉴别。McAb应用于淋巴白血病和恶性淋巴瘤的分类分型。近年来,培养细胞的免疫组化技术在鉴定细胞的种类、分化程度、表面抗原特点以及肿瘤结构成分改变等方面的研究均起到了积极作用。
细胞标本的取材有以下3种方法:
1.印片法主要应用于活组织检查标本和手术切除标本。新鲜标本以最大面积剖开,充分暴露病变区,将载玻片轻轻压于病变区,脱落的细胞便粘附在玻片上,立即浸入细胞固定液内5-10min,取出后自然干燥,低温保存备用。
优点是简便省时,细胞抗原保存较好。缺点是细胞分布不均匀,玻片上细胞重叠,影响标记效果。
2.穿刺吸取涂片法主要应用于实质器官的病变区,如肝、肾、肺、淋巴结、软组织等。用细针穿刺吸取病变区内液体成分,如穿刺液较少,可直接涂抹在载玻片上,力求细胞分布均匀。如穿刺液较多,细胞丰富,可用洗涤法:将穿刺液滴入盛有1-2ml Hanks液(RPMi 1640液)的试管内,轻轻搅拌,以500rpm低速离心5-10min后,弃上清液,将沉淀制成细胞悬液(浓度约2×106细胞/ml),吸取1滴于载玻片上,轻轻涂抹,待涂片略干即可固定。
该法穿刺吸取直接涂片的优点是操作简便,细胞形态保持较好。缺点是细胞分布不均匀。洗涂法片虽可弥补这一缺点,但操作复杂,细胞常常发生变形。
3.体液沉淀涂片法主要用于胸水、腹水、尿液、脑脊液等体液多、细胞少的标本。体液采取后,必须及时处理,更不宜加固定液。根据标本内细胞数量的多少选用不同的处理方法:①细胞数量极多者,可吸取少量液体直接涂在玻片上。②细胞数量较少者,可将液体自然沉淀,然后吸取5ml左右沉淀液,以1500rpm离心10min,弃上清液,将沉淀涂片,略干后固定备用。
如用细胞离心涂片器(Cytospin ),可将标本用上述离心沉淀法制成2×106细胞/ml的细胞悬液,吸取50μl加入涂片器内,离心后即制成分布均匀的细胞涂片,细胞分布在直径约6mm的小圆圈内,每个圆圈内的细胞数约105个(Danos,1976)。
培养细胞标本的取材可根据培养的细胞特性分别采取不同的方法。某些细胞有贴壁生长的特性,如纤维母细胞、粘液癌细胞等,只需将载玻片或盖玻片插入培养液内即可收集到理想的细胞标本。某些细胞只能在培养液中生长,可用上述体液沉淀离心涂片法处理。
制备细胞涂片应注意:①标本反复离心洗涤,细胞的粘附性降低,在免疫组化染色过程中容易脱片,因此,在制备涂片前载玻片上应涂粘附剂。②为节 省试剂和便于镜下观察和记数,应将细胞集中到直径0.6-1.0cm的圆圈内,细胞总数以105个为宜。③粘液丰富的标本,如痰液,胃液等,未经特殊处理,一般不宜作免疫组化标记。
(二)组织标本的取材
组织标本主要取之于活组织检查标本、手术切除标本、动物模型标本以及尸体解剖标本等。前三者均为新鲜组织,后者是机体死亡2h以上的组织,可能有不同程度的自溶,其抗原可能有变性消失,严重弥散现象,因此,尸检组织应尽快固定处理,以免影响免疫组化标记效果。但有些较稳定性抗原,如HBsAg、HBcAg等在尸检标本中,抗原显示仍较好。
组织标本的取材常常受到各种因素的影响,如各种内窥镜钳取的组织,常因过度挤压而变形,严重者组织结构被破坏。大组织标本病变分布广泛,抗原在组织中分布不均一,常出现人为的组织取材不准确。为了避免上述缺点,组织取材时应注意:①活检钳的刃口必须锋利,以免组织受挤压;②取材部位必须是主要病变区;③必须取病灶与正常组织交界区;④必要时取远距病灶区的正常组织作对照。
为充分保存组织的抗原性,标本离体后庆立即作处理,或立即速冻成冻块进行冰冻切片,或立即用固定液固定进行脱水、浸蜡、包埋、石蜡切片。如不能迅速制片,可贮存于液氮罐内或-70。C冰箱内备用。
二、细胞和组织的固定
(一)固定
为了更好的保持细胞和组织原有的形态结构,防止组织自溶,有必要对细胞和组织进行固定。固定的作用不仅是使细胞内蛋白质凝固,终止或抑制外源性和内源性酶活性,更重要的是最大限度的保存细胞和组织的抗原性,使水溶性抗原转变为非水溶性抗原,防止抗原弥散。
不同抗原,其稳定性也不相同,因而对固定剂的耐受性差异较大。如T淋巴细胞表面抗原属不稳定性抗原,对固定剂的耐受较差,抗原活性容易丧失。而HBsAg属稳定性抗原,其抗原活性很少受固定剂种类、固定时间、温度等因素的影响。
(二)固定剂
用于免疫组织化学的固定剂种类较多,性能各异,在固定半稳定性抗原时,尤其重视固定剂的选择,介绍如下。
1.醛类固定剂双功能交联剂,其作用是使组织之间相互交联,保存抗原于原位,其特点是对组织穿透性强,收缩性小。有人认为它对IgM、IgA、J链、K链和λ链的标记效果良好,背景清晰,是常用的固定剂。
(1)10%钙-福尔马林液(浓甲醛10ml,饱和碳酸钙90ml)。
(2)10%中性缓冲福尔马林液(浓甲醋10ml,0.01mol/l pH7.4 PBS 90ml)。
(3)4%多聚甲醛磷酸缓冲液pH7.4(多聚甲醛40g,0.1mol/L PBS液pH7.4500ml,两者混
合加热至60℃,搅拌并滴加1nNaOH至清晰为止,冷却后加PBS液至总量1000ml)。
(4)戊二醛-甲醛液(戊二醛1ml,浓甲醛10ml,蒸馏水加至100ml)。
戊二醛是二醛基化合物,交联结合力比甲醛大,Bullock认为交联过强,可出现组织改变和空间遮蔽现象,影响组织的抗原性。但McDonald等认为,该试剂用于PAP法免疫酶标记效果仍满意。
(5)甲醛升汞固定液(即B5固定液。浓甲醛10ml,氯化汞6g,醋酸钠1.25g,蒸馏水90ml)。
有人认为此固定液悬液是较理想的固定液,标记IgA、IgM、IgG等抗原效果良好。也有人认为它减弱细胞的抗原性,上皮细胞可产生非特异性荧光,故不宜用于免疫荧光标记。氯化汞是一种强蛋白凝固剂,但对组织穿透性弱,且使组织收缩,故与甲醛混合使用。
(6)醋酸-甲醛液(浓甲醛10ml,冰醋酸3ml,生理盐水加至100ml)。
Bullock等认为此液固定效果良好,组织可不经消化,胞浆IgA、IgG、IgM、IgD和K、λ、J链标记均呈阳性,且背景染色极淡。如标记IgG用PAP法第一抗体仅为甲醛升汞液的1/10。此液内醋酸既可防止组织收缩,又可暴露胞浆免疫球蛋白抗原决定簇。
(7)Bouin’s液。
该固定液为组织学、病理学常用固定剂之一,对组织穿透力较强而收缩性较小,比单独醛类固定更适合免疫组化染色。Kayhko认为用于标记B细胞的J链较好,但Bullock则认为它可导致Igg γ重链变性,故必需加大第一抗体的浓度。
(8)Zamboni’s液。
该固定液可用于电镜免疫细胞化学,对超微结构的保存优于纯甲醛,也适用于光镜免疫细胞化学研究。采用2.5%多聚甲醛和30%饱和苦味酸,更可增加对组织穿透力和固定效果,以保存更多的组织抗原。固定时间6-18h。
(9)PLP液(过碘酸盐-赖氨酸-多聚甲醛固定液)。
该固定剂适用于富含糖类的组织,对超微结构及许多抗原的抗原性保存较好。其机制是过碘酸氧化组织中的糖类形成醛基,通过赖氨酸的双价氧基与醛基结合,从而与糖形成交联。组织抗原大多数是由蛋白质和糖两部分构成,抗原决定簇位于蛋白部分,故该固定液有选择性地使糖类固定,既稳定缺的,又不影响其在组织中的位置(固定剂7-9配制法见附录1)。
2.非醛类固定剂Pe等人比较几种非醛类双功能试剂指出,碳化二亚胺、二甲基乙酰胺、二甲基辛酰亚胺、和对苯醌等均适用于多肽类激素的组织固定,单独使用时,边缘固定效应重,但与戊二醛或多聚甲醛混合使用,效果明显改善。
(1)碳化二亚胺(1-ethyl-3(3-dimethyl-aminopropyl)cardodiimi-HCI)液:2g溶于100ml0.01mol/L,pH7.4PBS中。此液宜用于标记多肽类激素的组织,对标记IgA、IgG效果不佳。
(2)碳二亚胺-戊二醛液(ECD-G液):配制法见附录一。
ECD-[1-ethyl-3(3-dimethyl-aminopropyl)carbodiimideHydrochloride],即乙基-二甲基氨基丙基碳亚胺盐酸盐,简称乙基-CDI。
该液常用于多肽类激素的固定,对酶等蛋白质固定效果良好,对细胞内抗原定位,超微结构保存好,是一种培养细胞电镜免疫细胞化学研究的良好固定剂。
(3)Zenker’s 液(重铬酸钾2.5g,氯化汞5g,硫酸钠1g,蒸馏水100ml,混合溶解后,临用时加水醋酸5ml)。
该固定液对免疫球蛋白染色最佳,固定时间约2-4h,染色前必须脱汞色素。
3.丙酮及醇类固定剂系最初免疫细胞化学染色的固定剂,其作用是沉淀蛋白质和糖,对组织穿透性很强,保存抗原的免疫活性较好。但醇类对低分子蛋白质、多肽及胞浆内蛋白质保存效果较差,解决的办法是和其它试剂混合使用,如加冰醋酸、乙醚、氯仿、甲醛等。
(1)Clarke氏改良剂(100%酒精95ml,冰醋酸5ml),用于冰冻切片的后固定。
(2)乙醚(或氯仿)与乙醇等量混合液。
Danos(1976)等认为其组织穿透性极强,即使涂片上富于过多的粘液,固定效果仍然良好,是理想的细胞固定液。
(3)AAF液:95%-100%酒精85ml,冰醋酸5ml,浓甲醛10ml。
(4)Carnoy氏液:100%酒精60ml,氯仿30ml,冰醋酸10ml,混合后4。C保存备用。
(5)Methacarn氏液:甲醇60ml,氯仿30ml,冰醋酸10ml,混合后4。C保存备用。
以上两种固定液适宜某些抗原,癌基因蛋白产物检测的 固定,P53抗癌基因蛋白产物,PC-NA等抗原的保存。
丙酮的组织穿透性和脱水性更强,常用于冰冻切片及细胞涂片的后固定,保存抗原性较好,平时4。C低温保存备用,临用时,只需将涂片或冰冻切片插入冷丙酮内5-10min,取出后自然干燥,贮存于低温冰箱备用。
以上介绍了免疫组织化学中常用的固定液,用于免疫组化的固定剂种类很多(见附录),不同的抗原和标本需经过反复试验,选用最佳固定液。不少学者认为,迄今尚无一种标准固定液可以用于各种不同的抗原固定。而且同一固定液固定的组织,免疫组化染色标记结果可截然不同,致使人们无所适从。选择最佳固定液标准是:①最好地保持细胞和组织的形态结构。②最大限度地保存抗原的免疫活性。一些含重金属的固定液在免疫组化技术中是禁用的。实际经验告诉我们,中性缓冲福尔马林(或多聚甲醛)是适应性较广泛的固定液,但固定时间不宜过长。必要时,可作多种固定液对比,从而选出理想的标准固定液。
固定组织时应注意:①应力求保持组织新鲜,勿使其干燥,尽快固定处理。②组织块不易过大过厚,必须小于2cm×1.5cm×0.3cm,尤其是组织块厚度必须控制在0.3cm以内。③固定液必须有足够的量,在体积上一般大于组织20倍以上,否则组织中心固定不良影响效果。④组织固定后应充分水洗,去除固定液造成的人为假象。
(三)固定方法
1.浸入法(Immersionmethod)将组织浸泡在固定液内,必要时可在低温(4。C)环境下进行,固定时间可根据抗原的稳定性以及固定液性质而定,一般在2-12h之间。
2.灌注法(Irrigationmethod)此法适用于动物实验研究。自左心室插入主动脉,先以krebs或生理盐水冲冼血液后,以泵、吊筒或50-100ml注射器注入固定液。置灌注动物于4。C冰箱内,次日取出脑组织或其它组织。外周组织一般在灌注后30min内取材,取组织置同一固定剂中浸入1-3h,然后修整组织块。有主张用冷固定剂(4。C)进行灌注。我们的体会是一般光镜下观察的标本,室温固定即可获满意效果。应该强调,保持组织新鲜是很重要的,据报告,肽类抗原活性在断绝血液供应后24h几乎完全丧失。
灌注法固定可使固定液迅速达到全身各组织。达到充分固定之目的。灌注冲洗还能排除红细胞内假过氧化物酶的干扰。浸入法主要用于活检和手术标本,以及其它不能进行灌注的组织固定。
三、组织切片技术
应用于光镜的免疫组织化学染色的切片厚度一般要求5μm左右,神经组织的研究要求切片厚度在20-100μm,有利于追踪神经纤维的走行。
1.冰冻切片是免疫组织化学染色中最常用的一种切片方法。其最突出的优点是能够较完好地保存多种抗原的免疫活性,尤其是细胞表面抗原更应采用冰冻切片。新鲜的组织及已固定的组织均可作冰冻切片。
冰冻时,组织中水份易形成冰晶,往往影响抗原定位。一般认为冰晶少而大时,影响较小,冰晶小而多时,对组织结构损害较大,在含水量较多的组织中上述现象更易发生。冰晶的大小与其生长速率成正比,而与成核率(形成速率)成反比,即冰晶形成的数量愈多则愈小,对组织结构影响愈严重。因此,应尽量降低冰晶的数量。Fish认为冰冻开始时,冰晶成核率较慢,以后逐渐增加,其临界温度为-33。C,从-30。C降至-43。C之间,成核率急剧增加达1018,然后再减慢。基于上述理论可采取以下措施减少冰晶的形成。
(1)速冻,使组织温度骤降,缩短从-33。C43。C的时间,减少冰晶的形成。其方法有二:
①干冰-丙酮(酒精)法:将150-200ml丙酮(酒精)装入小保温杯内,逐渐加入干冰,直至饱和呈粘稠状,再加干冰不再昌泡时,温度可达-70℃C,用一小烧杯(50-100ml)内装异戊烷约50ml,再将烧杯缓慢置入干冰丙酮(纯酒精)饱和液内,至异戊烷温度达-70℃时即可使用。将组织(大小为1cm×0.8cm×0.5cm)投入异戊烷内速冻30-60s后取出,或置恒冷箱内以备切片,或置-80℃低温冰箱内贮存。
②液氮法:将组织块平放于软塑瓶盖或特制小盒内(直径约2cm),如组织块小可适量加OCT包埋剂浸没组织,然后将特制小盒缓缓平放入盛有液氮的小杯内,当盒底部接触液氮时即开始气化沸腾,此时小盒保持原位切勿浸入液氮中,大约10-20s组织即迅速冰结成块。取出组织冰块立即置入-80℃冰箱贮存备用,或置入恒冷箱切片机冰冻切片。
(2)将组织置于20%-30%蔗糖溶液1~3天,利用高渗吸收组织中水分,减少组织含水量。
影响冰冻切片的因素较多,因此,技术难度较大,选择好的冰冻切片机是保证切片质量的关键。目前冰冻切片机有两类:
①恒慢冰冻切片机(Cryastat):为较理想的冰冻切片机,型号很多,但其基本结构是将切片机置于-30℃C低温密闭室内,故切片时不受外界温度和环境影响,可连续切薄片至2-4μm,完全能满足免疫组织化学标记要求。切片时,低温室内温度以-15℃~18℃为宜,温度过低组织易破碎,抗卷板的位置及角度要适当,载玻片附贴组织切片,切勿上下移动。
②开放式冰冻切片机:包括半导体致冷切片机和甲醇致冷切片以及老式的CO2、氯乙烷等冷冻切片机。切片时暴露空气中,温度不易控制,切片技术难度大,在高温季节 ,切片更加困难,且切片厚8~15μm,不易连续切片,但其优点是价廉,国内有生产。
冰冻切片后如不染色,必须吹干,贮存低温冰箱内,或进行短暂预固定后贮存冰箱保存。
2.石蜡切片其优点是组织结构保存良好,在病理和回顾性研究中有较大的实用价值,能切连续薄片,组织结构清晰,抗原定位准确。用于免疫组化技术的石蜡切片制备与常规制片略有不同:①脱水、透明等过程应在4℃。C下进行,以尽量减少组织抗原的损失。②组织块大小应限于2cm×1.5cm×0.2cm,使组织充分脱水、透明、浸蜡。③浸蜡、包埋过程中,石蜡应保持在60℃以下,以溶点低的软蜡最好(即低温石蜡包埋)。
组织块脱水、透明、浸蜡时间参考表1-3:
表 1-3 组织块处理时间表
| 1 | 70%乙醇 | 4℃ | 3~4h |
| 2 | 80%乙醇 | 4℃ | 3~4h |
| 3 | 90%乙醇 | 4℃ | 2~3h |
| 4 | 95%乙醇Ⅰ | 4℃ | 2~3h |
| 5 | 95%乙醇Ⅱ | 4℃ | 1~2h |
| 6 | 100%乙醇Ⅰ | 4℃ | 1.5h |
| 7 | 100%乙醇Ⅱ | 4℃ | 1.5h |
| 8 | 二甲苯Ⅰ | 4℃ | 0.5~1h |
| 9 | 二甲苯Ⅱ | 4℃ | 0.51~1h |
| 10 | 石蜡Ⅰ | 60℃ | 1h |
| 11 | 石蜡Ⅱ | 60℃ | 2h |
以上全过程为18-24h,也可在室温内使用自动脱水机代替。如组织块小,直径小于0.5cm,可用快速石蜡包埋切片,全过程只需4h左右。
石蜡切片为常规制片技术,切片机多为轮转式,切片厚度2~7μm,应用范围广,不影响抗体的穿透性,染色均匀一致。由于甲醛固定、有机熔剂和包埋剂对组抗原有一定的损害及遮蔽,使抗原特征发生改变。有人报告经蛋白酶消化,可以改善光镜免疫组化染色强度,常用的有胰蛋白酶、链霉蛋白酶及胃蛋白酶等消化法。石蜡切片应入37℃恒温箱过夜,这样烤片可减少染色中脱片现象。切片如需长期贮存,可存放于4℃冰箱内备用。
石蜡切片优点较多,但在制片过程中要经过酒精、二甲苯等有机溶剂处理,组织内抗原活性失去较多,有人采用冷冻干燥包埋法(Freeze drying embedding methed),可以保存组织内可溶性物质,防止蛋白变性和酶的失活,从而减少了抗原的丢失。该法是将新鲜组织低温速冻,利用冷冻干燥机(Freezing dryer)在真空、低温条件下排除组织内水分 ,然后用甲醛蒸气固定干燥的组织,最后将组织浸蜡、包埋、切片。此法可用于免疫荧光标记、免疫酶标记及放射自显影。
3.振动切片用振动切片机(Vibratotme),可以把新鲜组织(不固定不冰冻)切成厚片20~100μm,以漂浮法在反应板进行免疫组织化学染色,然后在解剖显微镜下检出免疫反应阳性部位,修整组织进行后固定,最后按电镜样品制备、脱水、包埋、超薄切片、染色观察等。组织不冰冻,无冰晶形成和组织抗原破坏,在免疫组化染色前避免了组织脱水、透明、包埋等步骤对抗原的损害,能较好地保留组织内脂溶性物质和细胞膜抗原,主要用于显示神经系统抗原分布研究。这种包埋前染色,尤其实用于免疫电镜观察。
4.塑料切片塑料包埋切片常用包埋剂有甲基丙烯酸盐类(Glycolmethacrylate, GMA)及环氧树脂类(Epon 812,618),其优点是可以同时作光镜和电镜检测,能相互对照所查抗原,定位准确。塑料包埋切片可切出比石蜡切片更薄的切片,光镜切片可薄至0.5~2um,故称半薄切片(Semithinsection)。GMA保存抗原较好,不与组织产生共聚合,但形态学结构欠佳。环氧树脂如Fpon和Araldite能较好地保存形态学结构,但在聚合过程中易和组织起作用,改变抗原结构。塑料包埋切片由于处理程序繁多,抗原活性易丢失。同时半薄切片进行免疫染色时,抗血清不易穿透树脂,因此,塑料切片主要用于免疫电镜的超微切片前定位。包埋前染色的标本,切片薄切片后不需染色,直接在相差显微镜下观察免疫反应部位呈黑点状,定位后进一步作超薄切片,这样,可以明显提高免疫电镜阳性检出率。
5.超薄切片电镜标本制作见第七章 ,应用超薄切片机(Ultrotome)进行切片。
6.碳蜡切片碳蜡(Carbowax),学名聚乙烯二醇(Polyethlene glycol, PEG)为水溶性蜡。根据其分子量不同,碳蜡有多种,如400、800、1000、1500、4000、6000等,用于组织包埋的有1500、4000两种,其熔点分别38℃C和52。C左右,常温下为固体石蜡状,加温熔化呈液体状。
本法的特点是组织固定水洗后,不需脱水透明可直接浸蜡包埋,且切片方法与常规石蜡切片相同。切下的组织片漂浮水面后自然展开,碳虹迅速深化,组织片即可展平,制片过程简单易行。
具体操作简述如下:组织块不宜过大,一般限定在1.5cm×1cm×0.1cm以内。①组织固定后充分水洗去除固定液,用滤纸吸干组织表面水。②将组织浸入碳蜡(1500)内,45℃30min。③再次浸入混合混合碳蜡液(1500和4000等量混合液),52℃30min。④浸入等量混合碳蜡液(或根据气温、湿度变化调整4000和5000混合比例,如气温高湿度大可以4000:1500=3:2混合,反之,则2:3混合)。⑤用等量混合碳蜡或调整混合碳蜡包埋成组织蜡块。⑥切片与石蜡切片相同。在操作过程中,碳蜡组织块应尽量避免与水或冰接触,贮存时应密封干燥冷藏。
本法优点是操作程序减少,时间缩短,组织不经有机溶剂损害、温度低、抗原性保存比石蜡切片好,组织结构清晰。缺点是夏季室温高时切片较困难,连续切片不如石蜡切片顺利;由于碳蜡有强吸湿性,不易长期保存。
7.玻片处理和涂胶在免疫组化染色过程中由于各种原因常造成标本(细胞制片和组织切片)脱片现象,影响了工作质量和速度。一般采取两种方法即可防止脱片现象出现。
(1)载玻片和盖玻片处理:新载玻片上有油污,必须经过清洁液浸泡12~24h,流水充分漂洗后用蒸馏水清洗5遍以上,浸泡在95%酒精内2h,用绸布擦干或用红外线烤箱烤干均可,贮放于玻片盒内备用。盖玻片很薄,以上处理程序必须缩短,清洁液浸泡只需2h,流水冲洗注意勿损伤玻片等。
(2)载玻片上涂粘附剂:粘附剂种类较多,常用的有以上几种:①树脂胶(Resin glue)又称白色乳胶,为木工用,有进口,国产之分,有人认为进口白色树脂胶较稳定,我们认为国产白乳胶(聚醋酸乙烯乳液J-北京产)质量尚稳定。使用浓度为1%~2%,以蒸馏水稀释即可。②甘油明胶,混匀后涂在载玻片上,切片贴附后置有副醛的干燥器内,加热80℃1h。③甲醛明胶, 混合后即可涂片。④铬明矾明胶, 混合后即可涂片,置37℃温箱烤干。⑤多聚赖氨酸液:多聚赖氨酸0.1g,加蒸馏水10ml,混合后即可涂片。此液不宜多配制。⑥3-Amino propy Eri-ethoxy Silame(APES)。
粘附剂2~5配制法见附录一。
参考文献
1.Bullock CR, etal . Techniques in immunocytochemistry, Vol 1. London New York: Academic Press,1982
2.Coleman DV, et al.Immunoperoxidase staining tumor marker distribution studies in cytologicspecimens. Acta Cytologica, 1981:25:205~206
3.Jacobsen M, etal .The effectof fixation and trypsinization on the immunohistochemical demonstration ofintracellular immunoglobulin in paraffin embedded material .Acto Path.Microbiol . Scand . ,Sect. A, 1980;88:369~376
4. Kayhko?K. Optimal fixation ofthe immunoperoxidase identification of human J chain from tissue sections.Histochemistry, 1980;70:23-27
5.Leathem A, et al .Fixation andimmunohistochemistry of lymphoid tissue.J. Clin Path, 1980;33:1010-1012
6.Martin SE,et al Immunologicmethods in cytology:definitive diagnosis of non-Hodgkin’s lymphomas usingimmunologic markers for T-and B-cells .Am . Clin . Path.,1984;82:666~673
7.Mason DY,et al, Technicalaspects of lymphoma immunohistology.J. Histochem. Cytochem., 1980;28:731-745
8.Montero C.Immunocytochemistry.2nd ed. New York; John Wiley and Sons, 1979
9.Walts AE, et al.SPecial tumormarkers in diagnostic cytology:immunoperoxidase of carinoembryonicantigen,lysozyme and other tissue antigen in effusions, washes and aspirates.Acta Cytologica, 1983;27:408~416
10.Wordinger RJ, et al. Manual ofimmunoperoxidase techniques. Chicago; Americal Society of Pathologists Press,1983
11.施达仁,等。B淋巴细胞双PAP法免疫定位方法学的探讨及其在恶性淋巴瘤诊断上的应用。肿瘤,1982;2:11-14
12.Nuovo GJ.Comparison of Bouin solution and buffered formalin fixation on the detectionrate by in hybridization of human papillomavins DND in qenital tract lesions.
第二章 抗原和抗体的制备
第一节 抗原的制备(缺)
第二节 抗体的制备
一、抗血清的制备
有了质量好的抗原,还必须选择适当的免疫途径,才能产生质量好(特异性强和效价高)的抗体。
(一)用于免疫的动物
作免疫用的动物有哺乳类和禽类,主要为羊、马、家兔、猴、猪、豚鼠、鸡等,实验室常用者为家兔、山羊和豚鼠等。动物种类的选择主要根据抗原的生物学特性和所要获得抗血清数量,如一般制备抗r-免疫球蛋白抗血清,多用家兔和山羊,因动物反应良好,而且能够提供足够数量的血清,用于免疫的动物应适龄,健壮,无感染性疾患,最好为///雄性,此外还需十分注意动物的饲养,以消除动物的个体差异以及在免疫过程中死亡的影响。若用兔,最好用纯种新西兰兔,一组三只,兔的体重以2~3kg为宜。
(二)免疫途径
免疫途径有多种多样,如静脉内、腹腔内、肌肉内、皮内、皮下、淋巴结内注射等,一般常用皮下或背部多点皮内注射,每点注射0.1ml左右。途径的选择决定于抗原的生物学特性和理化特性,如激素、酶、毒素等生物学活性抗原,一般不宜采用静脉注射。
(三)佐剂
由于不同个体对同一抗原的反应性不同,而且不同抗原产生免疫反应的能力也有强有弱,因此常常在注射抗原的同时,加入能增强抗原的抗原性物质,以刺激机体产生较强的免疫反应,这种物质称为免疫佐剂。
佐剂除了延长抗原在体内的存留时间,增加抗原刺激作用外,更主要的是,它能刺激网状内皮系统,使参与免疫反应的免疫活性细胞增多,促进T细胞与B细胞的相互作用,从而增强机体对抗原的细胞免疫和抗体的产生。
常用的佐剂是福氏佐剂(Freund adjuvant),其成分通常是羊毛脂1份、石腊油5份,羊毛脂与石腊油的比例,视需要可调整为1:2~9(V/V),这是不完全福氏佐剂,在每毫升不完全佐剂加入1~20mg卡介苗就成为完全佐剂。
配制方法:按比例将羊毛脂与石蜡油置容器内,用超声波使之混匀,高压灭菌,置4℃下保存备用。免疫前取等容积完全或不完全佐剂与免疫原溶液混合,用振荡器混匀成乳状,也可以在免疫前取需要量佐剂置乳钵中研磨,均匀后再边磨边滴加入等容积抗原液(其中加卡介苗3~4mg/ml或不加),加完后再继续研磨成乳剂,滴于冰水上5~10min内完全不扩散为止。为避免损失抗原,亦可用一注射器装抗原液,另一注射器装佐剂,二者以聚乙烯塑料管连接,然后二者来回反复抽吸,约数十分钟后即能完全乳化。检查合格后即以其中一注射器作注射用。
(四)免疫方法
抗原剂量,首次剂量为300~500μg,加强免疫的剂量约为首次剂量为1/4左右。每2~3周加强免疫一次。加强免疫时用不完全佐剂,首次免疫时皮下注射百日咳疫苗0.5ml,加强免疫时不必注射百日咳疫苗。
在第2次加强免疫后2周,从耳缘静脉取2~3ml血,制备血清,检测抗体效价(见后)。如未达到预期效价,需再进行加强免疫,直到满意时为止(图2-3)。当抗体效价达到预期水平时,即可放血制备抗血清。
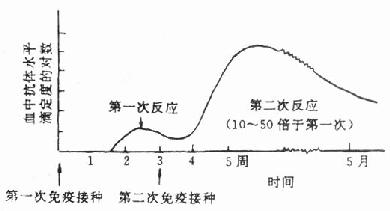
图2-3 抗体反应
(五)抗血清的采集与保存
家兔是最常用以产生抗体的动物,因此这里主要讨论兔血的收集。羊等较大动物以颈静脉、动脉取血,鼠等小动物取血可参阅有关资料。取兔血有两种方法,一是耳缘静脉或耳动脉放血,一是颈动脉入血,也可心脏采血。取动脉或静脉放血时,将兔放入一个特造的木匣或笼内,耳露于箱(笼)外,也可由另一人捉住兔身。剪去耳缘的毛,用少许二甲苯涂抹耳廓,30s后,耳血管扩张、充血。用手轻拉耳尖,以单面剃须刀或尖的手术刀片,快速切开动脉或静脉,血液即流出,每次可收集30~40ml 。然后用棉球压迫止血,凝血后洗去二甲苯。二星期后,可在另一耳放血。此法可反复多次放血。颈动脉放血时,将兔仰卧,固定于兔台,剪去颈部的毛,切开皮肤,暴露颈动脉,插管,放血。放血过程中要严格按无菌要求进行。
收集的血液置于室温下1h左右,凝固后,置4℃下,过夜(切勿冰冻)析出血清,离心,4000rpm,10min。在无菌条件,吸出血清,分装(0.05~0.2ml),贮于-40℃以下冰箱,或冻干后贮存于4。C冰箱保存。
(六)抗血清质量的评价
在免疫期间,不仅各个不同的动物,而且同一动物在不同的时间内抗血清效价、特异性、亲合力等都可能发生变化,因而必须经常地采血测试。只有在对抗血清的效价、特异性、亲合力等方面作彻底的评价后,才可使用所取得的抗血清。
1.效价 抗血清的效价,就是指血清中所含抗体的浓度或含量。效价测定的方法常用的是放射免疫法,此法对所有的抗体均适用。某些由大分子(如蛋白类)抗原所产生的抗体,可用双扩散等方法测定。前者测定的效价极为精确。而后者则粗糙得多。
(1)放射免疫法: 以不同稀释度的抗血清与优质标记抗原混合,孵育24h后,测定其结合率。通常以结合率为50%的血清稀度和为效价。如某抗血清的结合率为50%时的稀释度为1:15000,则该血清的效价就是1:15000。抗血清的效价,除由抗血清本身的性质决定外,还受标记抗原的质量、孵育时间,所用稀释液的成分及其pH等因素的影响,在工作中必须引起注意。(测定方法见第8章 )
(2)双向扩散法:利用大分子抗原和抗体在琼脂平板上扩散,两者在相交处产生沉淀线,以观察和判断抗血清中是否有抗体及其浓度。
球脂板的制备:100ml pH7.1 的磷酸盐缓冲液加到15g的琼脂内,于水浴中加温,搅拌,使琼脂完全溶解,趁热用纱布过滤,待溶液冷却到65℃左右时,加入叠氮钠(NaN3),使其在溶液中的浓度为0.1%。用移液管把琼脂放在干净平皿或玻片上,约3mm厚,待其冷却,完全凝固后,用打孔器打孔(图2-4)。中央孔内加适量抗原(容量为50μl),周围各孔内分别加入50μl 1:2、1:4、1:8、1:16、1:32及不稀释的抗血清,37℃下孵育24h,观察有无沉淀线产生,以判断血清的稀释度(图2-5)。
图2-4 双向扩散模型
图2-5 免疫扩散试验
2.特异性测定 抗血清的特异性或称专一性是指抗血清对相应的抗原及近似的抗原物质的识别能力。特异性好就是抗血清的识别能力强。通常,特异性是以交叉反应率来表示的。交叉反应率低,表示抗血清的特异性好,反之则特异性差。交叉反应率一般是用竞争抑制曲线来判断的。以不同浓度的抗原和近似抗原物质分别做竞争抑制曲线,计算各自的结合率(B/T或B/B0),求出各自在IC50时的浓度,按下列公式计算交叉反应率。
S=y/Z×100%
S:交叉反应率,y:IC50时抗原浓度,Z:IC50时近似抗原物质的浓度。
如某抗原的IC50浓度为90Pg/管,而一些近似抗原物质IC50浓度几乎是无限大,可以说这一抗血清与其它抗原物质的交叉反应率近似零,即无交叉反应,该抗血清的特异性是好的。
3.亲合力 在免疫学中, 亲合力是指抗体与结合抗原体的活度或牢固度。抗体与抗原结合疏松,结合后会迅速解离,称为亲合力低,反之,亲合力高。亲合力的高低是由抗原分子的大小,抗体分子的结合位点与抗原的决定基之间的立体结构型的合适程度决定的。亲合力常以亲合常数K表示。K的单位是升/摩尔(L/mol)。在RIA中,K是该抗血清能达到的最小检出量(灵敏度)的倒数,K=1/[H],[H]是最小检出量,通常,K的范围在108~1012L/mol之间,也有高达1014L/mol的。
计算亲合常数的方法20余种,计算出的K都不能真实反映实验情况,只能作为参考。
(七)免疫失败的可能原因及应采取的措施
有时不能获得满意的抗血清,可从下列几方面找原因,并改进之。
(1)免疫动物的种属及品系是否合适,可考虑改变动物的种属或品系,或扩大免疫动物的数量。
(2)抗原质量是否良好,可改用其它厂家的产品或改用同一厂家的其它批号,也可考虑改变抗原分子的部分结构,或改进提取方法。
(3)制备的免疫原是否符合要求,可从偶联剂,载体、抗原或载体的比例、反应时间等多方面去考虑,并加以改进。
(4)所用的佐剂是否合适,乳化是否完全,可改用其它佐剂,或加强乳化。
(5)免疫的方法、剂量,加强免疫的间隔时间和次数,免疫的途径是否合适。
(6)动物的饲养是否得当,如营养(饲料、饮水)、环境卫生(通风、采光、温度)是否符合要求,动物的健康情况是否良好等。
二、单克隆抗体的制备
1975年Kohler和Milstein发现将小鼠骨髓瘤细胞与和绵羊红细胞免疫的小鼠脾细胞进行融合,形成的杂交瘤细胞既可产生抗体,又可无性繁殖,从而创立了单克隆抗体杂交瘤技术。这一技术上的突破使血清学的研究进入了一个高度精确的新纪元。
免疫细胞化学的 技术关键之一是制备特异性强、亲合力大、滴度高的特异性抗体,由于每种抗原都有几个抗原决定簇,用它免疫动物将产生对各个决定簇的抗体,即多克隆抗体。单克隆抗体则是由一个产生抗体的细胞与一个骨髓瘤细胞融合而形成的杂交廇细胞经无性繁殖而来的细胞群所产生的,所以它的免疫球蛋白属同一类型,质地纯一,而且它是针对某一抗原决定簇的,因此特异性强,亲合性也一致。单克隆抗体(McAb)的特性和常规血清抗体的特性比较见2-3。
表2—3 单克隆抗体(McAb)和常规免疫血清抗体的特性比较
| 项目 | 常规免疫血清抗体 | McAb |
| 抗体产生细胞 | 多克隆性 | 单克隆性 |
| 抗体的结合力 | 特异性识别多种抗原决定簇 | 特异性识别单一抗原决定簇 |
| 免疫球蛋白类别及亚类 | 不均一性,质地混杂 | 同一类属,质地纯一 |
| 特异性与亲合力 | 批与批之间不同 | 特异性高,抗体均一 |
| 有效抗体含量 | 0.01~0.1mg/ml(小鼠腹水) | 0.5~5.0mg/ml(小鼠腹水) 0.5~10.0μg/ml(培养物上清液) |
| 用于常规免疫学实验 | 可用 | 单抗组合应用 |
| 抗原抗体形成格子结构(沉淀反应) | 容易形成 | 一般难形成 |
| 抗原抗体反应 | 抗体混杂,形成2分子反应困难,不可逆 | 可形成2分子反应,可逆 |
单克隆抗体的制备方法如下。
(一)动物的选择与免疫
1.动物的选择 纯种BALB/C小鼠,较温顺,离窝的活动范围小,体弱,食量及排污较小,一般环境洁净的实验室均能饲养成活。目前开展杂交瘤技术的实验室多选用纯种BALA/C小鼠。
2.免疫方案 选择合适的免疫方案对于细胞融合杂交的成功,获得高质量的McAb至关重要。一般在融合前两个月左右根据确立免疫方案开始初次免疫,免疫方案应根据抗原的特性不同而定。
(1)可溶性抗原免疫原性较弱,一般要加佐剂,半抗原应先制备免疫原,再加佐剂。常用佐剂:福氏完全佐剂、福氏不完全佐剂。
初次免疫 抗原1~50μg加福氏完全佐剂皮下多点注射或脾内注射(一般0.8~1ml,0.2ml/点)
↓3周后
第二次免疫 剂量同上,加福氏不完全佐剂皮下或ip(腹腔内注射)(ip剂量不宜超过0.5ml)
↓3周后
第三次免疫 剂量同一,不加佐剂,ip(5~7天后采血测其效价)
↓2~3周
加强免疫,剂量50~500μg为宜,ip或iv(静脉内注射)
↓3天后
取脾融合
目前,用于可溶性抗原(特别是一些弱抗原)的免疫方案也不断有所更新,如:①将可溶性抗原颗粒化或固相化,一方面增强了抗原的免疫原性,另一方面可降低抗原的使用量。②改变抗原注入的途径,基础免疫可直接采用脾内注射。③使用细胞因子作为佐剂,提高机体的免疫应答水平,增强免疫细胞对抗原的反应性。
(2)颗粒抗原免疫性强,不加佐剂就可获得很好的免疫效果。以细胞性抗原为例,免疫时要求抗原量为1~2×107个细胞。
初次免疫 1×107/0.5ml ip
↓2~3周后
第二次免疫 1×107/0.5ml ip
↓3周后
加强免疫(融合前三天)1×107/0.5ml ip或iv
↓
取脾融合
(二)细胞融合
1.细胞融合前准备
(1)骨髓瘤细胞系的选择:骨髓瘤细胞应和免疫动物属于同一品系,这样杂交融合率高,也便于接种杂交瘤在同一品系小鼠腹腔内产生大量McAb。常用的骨髓瘤细胞系见表2-4。
表2-4用于融合试验的主要骨髓瘤细胞系
| 名 称 | 来 源 | 耐 受 药 物 | Ig链 |
| H L | |||
| P3/X63-Ag8(X63) | BALB/C骨髓瘤MOPC-21 | 8-氮鸟嘌呤 | r1 K |
| P3/X63-Ag8.653(X63-Ag8.653) | P3/X63-Ag8 | 8-氮鸟嘌呤 | - - |
| P3/NSI-1-Ag4-1(NS-1) | P3/X63-Ag8 | 8-氮鸟嘌呤 | - K(不分泌型) |
| P3/X63-Ag8.Ul(P3Ul) | (X63×BALB/C脾细胞)杂交瘤 | 8-氮鸟嘌呤 | - - |
| SP2/0-Ag14(SP2/0) | (X63×BALB/C脾细胞)杂交瘤 | 8-氮鸟嘌呤 | - - |
| F0 | BALB/C骨髓瘤 | 8-氮鸟嘌呤 | - - |
| S194/5.XXO.BU.1 | P3/X63-Ag8 | 5-溴脱氧尿嘧啶核苷 | - - |
| MPC11-45.6TG1.7 | BALB/C骨髓瘤MPC-11 | 6-巯鸟嘌呤 | r2b K |
| 210.RCY3.Ag1.2.3 | LOU大鼠骨髓瘤R210 | 8-氮鸟嘌呤 | - K |
| GM15006TG-A12 | 人骨髓瘤GM1500 | 6-巯鸟嘌呤 | r1 K |
| U-266AR | 人骨髓瘤U-266 | 8-氮鸟嘌呤 | ελ |
骨髓瘤细胞的培养可用一般的培养液,如RPMI1640,DMEM培养基。小牛血清的浓度一般在10%~20%,细胞浓度以104~5×105/ml为宜,最大浓度不得超过106/ml。当细胞处于对数生长的中期时,可按1:3~1:10的比例传代。每3~5天传代一次。细胞在传代过程中,部分细胞可能有返祖现象,应定期用8-氮鸟嘌呤进行处理,使生存的细胞对HAT呈均一的敏感性。
(2)饲养细胞:在组织培养中,单个或少数分散的细胞不易生长繁殖,若加入其它活细胞,则可促进这些细胞生长繁殖,所加入的这种细胞数被称为饲养细胞。在制备McAb的过程中,许多环节 需要加饲养细胞,如在杂交瘤细胞筛选、克隆化和扩大培养过程中,加入饲养细胞是十分必要的。常用的饲养细胞有:小鼠腹腔巨噬细胞(较为常用)、小鼠脾脏细胞或胸腺细胞。也有人用小鼠成纤维细胞系3T3经放射线照射后作为饲养细胞。饲养细胞的量为一般为2×104或105细胞/孔。
2.细胞融合的步骤
(1)制备饲养细胞层:一般选用小鼠腹腔巨噬细胞。
与免疫小鼠相同品系的小鼠,常用BALB/C小鼠,6~10周龄
↓
拉颈 处死,浸泡在75%酒精内,3~5min
↓
用无菌剪刀剪开皮肤,暴露腹膜
↓
用无菌注射器注入5~6ml预冷的培养液(严禁刺破肠管)
↓
反复冲洗,吸出冲洗液
↓
冲洗液放入10ml离心管,1200rpm/分离5~6min
↓
用20%小牛血清(NCS)或胎牛血清(FCS)的培养液混悬,调整细胞数至1×105/ml
↓
加入96孔板,100μl/孔
↓
放入37。c CO2孵箱培养
(2)制备免疫脾细胞
最后一次加强免疫3天后小鼠拉颈处死
↓
无菌取脾脏,培养液洗 一次
↓
脾脏研碎,过不锈钢筛网
↓
离心,细胞用培养液洗2次
↓
计数
↓
取108脾淋巴细胞悬液备用
(3)制备骨髓瘤细胞
取对数生长骨髓瘤细胞离心
↓
用无血清培养液洗2次
↓
计数,取得×107细胞备用
(4)融合
①将骨髓瘤细胞与脾细胞按1:10或1:5的比例混合在一起,在50ml离心管中用无血清不完全培养液洗1次,离心,1200rpm,8min;弃上清,用吸管吸净残留液体,以免影响聚乙二醇(PEG)浓度。轻轻弹击离心管底,使细胞沉淀略松动。
②90s内加入37℃预温的1ml 45%PEG(分子量4000)溶液,边加边轻微摇动。37℃水浴作用90s。
③加37。C预温的不完全培养液以终止PEG作用,每隔2min分别加入1ml、2ml、3ml、4ml、5ml和6ml。
④离心,800rpm, 6min。
⑤充上清,用含20%小牛血清HAT选择培养液重悬。
⑥将上述细胞,加到已有饲养细胞层的96孔板内,每孔加100μl。一般一个免疫脾脏可接种4块96孔板。
⑦将培养板置37℃、5%CO2培养箱中培养。
(三)选择杂交瘤细胞及抗体检测
1.HAT选择杂交瘤细胞 脾细胞和骨髓瘤细胞经PEG处理后,形成多种细胞的混合体,只有脾细胞与骨髓细胞形成的杂交瘤细胞才有意义。在HAT选择培养液中培养时,由于骨髓瘤细胞缺乏胸苷激酶或次黄嘌呤鸟嘌呤核糖转移酶,故不能生长繁殖,而杂交瘤细胞具有上述两种酶,在HAT选择培养液可以生长繁殖。
在用HAT选择培养1~2天内,将有大量瘤细胞死亡,3~4天后瘤细胞消失,杂交细胞形成小集落,HAT选择培养液维持7~10天后应换用HT培养液,再维持2周,改用一般培养液。在上述选择培养期间,杂交瘤细胞布满孔底1/10面积时,即可开始检测特异性抗体,筛选出所需要的杂交瘤细胞系。在选择培养期间,一般每2~3天换一半培养液。
2.抗体的检测 检测抗体的方法应根据抗原的性质、抗体的类型不同,选择不同的筛选方法,一般以快速、简便、特异、敏感的方法为原则。
常用的方法有:(1)放射免疫测定(RIA)可用于可溶性抗原、细胞McAb的检测。(2)酶联免疫吸附试验(ELISA)可用于可溶性抗原、细胞和病毒等McAb的检测。(3)免疫荧光试验适合于细胞表面抗原的McAb的检测。(4)其它如间接血凝试验、细胞毒性试验、旋转粘附双层吸附试验等。
(四)杂交瘤的克隆化
杂交瘤克隆化一般是指将抗体阳性孔进行克隆化。因为经过HAT筛选后的杂交瘤克隆不能保证一个孔内只有一个克隆。在实际工作中,可能会有数个甚至更多的克隆,可能包括抗体分泌细胞、抗体非分泌细胞、所需要的抗体(特异性抗体)分泌细胞和其它无关抗体的分泌细胞。要想将这些细胞彼此分开就需要克隆化。克隆化的原则是,对于检测抗体阳性的杂交克隆尽早进行克隆化,否则抗体分泌的细胞会被抗体非分泌的细胞所抑制,因为抗体非分泌细胞的生长速度比抗体分泌的细胞生长速度快,二者竞争的结果会使抗体分泌的细胞丢失。即使克隆化过的杂交瘤细胞也需要定期的再克隆,以防止杂交瘤细胞的突变或染色体丢失,从而丧失产生抗体的能力。
克隆化的方法很多,最常用的是有限稀释法和软琼脂平板法。
1.有限稀释法克隆
(1)克隆前1天制备饲养细胞层(同细胞融合)。
2)将要克隆的杂交瘤细胞从培养孔内轻轻吹干,计数。
(3)调整细胞为3~10个细胞/ml。
(4)取头天准备的饲养细胞层的细胞培养板,每孔加入稀释细胞100μl。孵育于37℃、5%CO2孵箱中。
(5)在第7天换液,以后每2~3天换液1次。
(6)8~9天可见 细胞克隆形成,及时检测抗体活性。
(7)将阳性孔的细胞移至24孔板中扩大培养。
(8)每个克隆应尽快冻存。
2.软琼脂培养法克隆
(1)软琼脂的配制:含有20%NCS(小牛血清)的2倍浓缩的RPMI1640。
①1%琼脂水溶液:高压灭菌,42℃预热。
②0.5%琼脂:由1份1%琼脂加1份含20%小牛血清的2倍浓缩的RPMI1640配制而成。置42℃保温。
(2)用上述0.5%琼脂液(含有饲养细胞)15ml倾注于直径为9cm的平皿中,在室温中待凝固后作为基底层备用。
(3)按100/ml,500/ml或5000/ml等浓度配制需克隆的细胞悬液。
(4)1ml 0.5%琼脂液(42℃预热)在室温中分别与1ml不同浓度的细胞悬液相混合。
(5)混匀后立倾注于琼脂基底层上,在室温中10min,使其凝固,孵育于37℃、5%CO2孵箱中。
(6)4~5天 后即可见针尖大小白色克隆,7~10天后,直接移种至含饲养细胞的24孔中进行培养。
(7)检测抗体,扩大培养,必要是地再克隆化。
(五)杂交瘤细胞的冻存与复苏
1.杂交瘤细胞的冻存 及时冻存原始孔的杂交瘤细胞每次克隆化得到的亚克隆细胞是十分重要的。因为在没有建立一个稳定分泌抗体的细胞系的时候,细胞的培养过程中随时可能发生细胞的污染、分泌抗体能力的丧失等等。如果没有原始细胞的冻存,则可因上述意外而前功尽弃。
杂交瘤细胞的冻存方法同其他细胞系的冻存方法一样,原则上细胞应在每支安瓿含1×106以上,但对原始孔的杂交瘤细胞可以因培养环境不同而改变,在24孔培养板中培养,当长满孔底时,一孔就可以装一支安瓿冻存。
细胞冻存液:50%小牛血清;40%不完全培养液;10%DMSO(二甲基亚砜)。
冻存液最好预冷,操作动作轻柔、迅速。冻存时从室温可立即降至0℃后放入-70℃超低温冰箱,次日转入液氮中。也可用细胞冻存装置进行冻存。冻存细胞要定期复苏,检查细胞的活性和分泌抗体的稳定性,在液氮中细胞可保存数年或更长时间。
2.细胞复苏方法 将玻璃安瓿自液氮中小心取出,放37℃水浴中,在1min内使冻存的细胞解冻,将细胞用完全培养液洗涤两次,然后移入头天已制备好的饲养层细胞的培养瓶内,置37℃、5%CO2孵箱中培养,当细胞形成集落时,检测抗体活性。
(六)单克隆抗体的大量生产
大量生产单克隆抗体的方法主要有两种:
(1)体外使用旋转培养管大量培养杂交瘤细胞,从一清液中获取单克隆抗体。但此方法产量低,一般培养液内抗体含量为10~60μg/ml,如果大量生产,费用较高。
(2)体内接种杂交瘤细胞,制备腹水或血清。
①实体瘤法:对数生长期的杂交瘤细胞按1~3×107/ml接种于小鼠背部皮下,每处注射0.2ml,共2~4点。待肿瘤达到一定大小后(一般10~20天)则可采血,从血清中获得单克隆 抗体的含量可达到1~10mg/ml。但采血量有限。
②腹水的制备:常规是先腹腔注射0.5mlPristane (降植烷)或液体石蜡于BALB/C鼠,1~2周后腹腔注射1×106个杂交瘤细胞,接种细胞7~10天后可产生腹水,密切观察动物的健康状况与腹水征象,待腹水尽可能多,而小鼠频于死亡之前,处死小鼠,用滴管将腹水吸入试管中,一般一只小鼠可获5~10ml腹水。也可用注射器抽提腹水,可反复收集数次。腹水中单克隆抗体含量可达到5~10mg/ml,这是目前最常用的方法,还可将腹水中细胞冻存起来,复苏后转种小鼠腹腔则产生腹水块、量多。
(七)单克隆抗体的鉴定
对制备的McAb进行系统的鉴定是十分必要的,应做下述几个方面的鉴定:
1.抗体特异性的鉴定 除用免疫原(抗原)进行抗体的检测外,还应该用与其抗原成分相关的其它抗原进行交叉试验,方法可用ELISA、IFA法。例如:①制备抗黑色素瘤细胞的McAb,除用黑色素瘤细胞反应外,还应该用其它脏器的肿瘤细胞和正常细胞进行交叉反应,以便挑选肿瘤特异性或肿瘤相关抗原的单克隆抗体。②制备抗重组的细胞因子的单克隆抗体,应首先考虑是否与表达菌株的蛋白有交叉反应,其次是与其它细胞因子间有无交叉。
2.McAb的Ig类与亚类的鉴定一般在用酶标或荧光素标记的第二抗体进行筛选时已经基本上确定了抗体的Ig类型。如果用的是酶标或荧光素标记的兔抗鼠IgG或IgM,则检测出来的抗体一般是IgG类或IgM类。至于亚类则需要用标准抗亚类血清系统作双扩或夹心ELISA来确定。在作双扩试验时,如加入适量的PEG(3%),更有利于沉淀线的形成。
3.McAb中和活性的鉴定 用动物或细胞的保护实验来确定McAb的生物学活性。例如,如果确定抗病毒McAb的中和活性,则可用抗体和病毒同时接种于易感的动物或敏感的细胞,来观察动物或细胞是否得到抗体的保护。
4.McAb识别抗原表位的鉴定用竞争结合试验,测相加指数的方法,测定McAb所识别抗原位点,来确定McAb的识别的表位是否相同。
5.McAb亲合力的鉴定 用ELISA或RIA竞争结合试验来确定McAb与相应抗原结合的亲合力。
第三节 抗体的提取与纯化(缺)
第三章 免疫荧光细胞化学技术
免疫荧光细胞化学是现代生物学和医学中广泛应用的技术之一,是由Coons等(1950)建立,经过近43年的发展,免疫荧光技术与形态学技术相结合发展成免疫荧光细胞(或组织)化学。它与亲合化学技术如葡萄球菌A蛋白(SPA)、生物素与卵白素、植物血凝素(ConA等)相结合拓宽了领域;与激光技术、电子计算机和扫描电视等技术结合发展为定量免疫荧光细胞化学技术;荧光激活细胞分类器(FACS)的应用使免疫荧光细胞化学技术发展到更高的阶段,开创了免疫荧光技术的新领域。细胞显微分光亮度计与与图像分析仪的结合使免疫荧光组织化学的定量检测更加准确。在免疫荧光细胞化学中应用单克隆抗体日益增多,将会不断提高特异性、敏感性和应用范围。激光扫描等聚集显微镜的应用又开创了新的发展时代。
由于免疫荧光细胞化学的特异性,快速性和在细胞和分子水平定位的敏感性与准确性,在免疫学、微生物学、细胞和组织学、病理学、肿瘤学以及临床检验学等生物学和医学许多方面得到广泛应用,日益发挥重要的作用。
第一节 免疫荧光细胞化学的原理
免疫荧光细胞化学是根据抗原抗体反应的原理,先将已知的抗原或抗体标记上荧光素制成荧光标记物,再用这种荧光抗体(或抗原)作为分子探针检查细胞或组织内的相应抗原(或抗体)。在细胞或组织中形成的抗原抗体复合物上含有荧光素,利用荧光显微镜观察标本,荧光素受激发光的照射而发出明亮的荧光(黄绿色或桔红色),可以看见荧光所在的细胞或组织,从而确定抗原或抗体的性质、定位,以及利用定量技术测定含量(图3-1)。
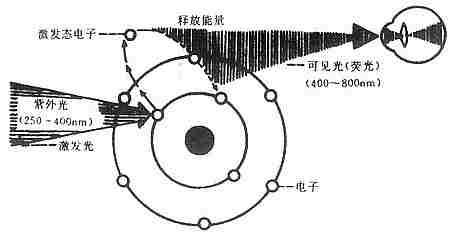
图3-1 紫外光激发荧光物质放射荧光示意图
用荧光抗体示踪或检查相应抗原的方法称荧光抗体法;用已知的荧光抗原标记物示踪或检查相应抗体的方法称荧光抗原法。这两种方法总称免疫荧光技术,以荧光抗体方法较常用。用免疫荧光技术显示和检查细胞或组织内抗原或半抗原物质等方法称为免疫荧光细胞(或组织)化学技术。
免疫荧光细胞化学分直接法、夹心法、间接法和补体法。
一、直接法
1.检查抗原法 这是最早的方法,用已知特异性抗体与荧光素结合,制成荧光特异性抗体,直接与细胞或组织中相应抗原结合,在荧光显微镜下即可见抗原存在部位呈现特异性荧光。此法很特异和简便,但一种荧光抗体只能检查一种抗原,敏感性较差(图3-2)。
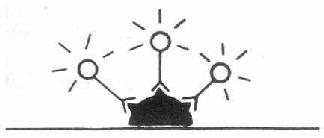
图3-2 直接法
2.检查抗体法 将抗原标记上荧光素,即为荧光抗原,用此荧光抗原与细胞或组织内相应抗体反应,而将抗体定位检测出来。
二、间接法
1.检查抗体法(夹心法)此法是先用特异性抗原与细胞或组织内抗体反应,再用此抗原的特异性荧光抗体与结合在细胞内抗体上的抗原相结合,抗原夹在细胞抗体与荧光抗体之间,故称夹心法。
2.检查抗体法用已知抗原细胞或组织标本的切片,加上待检血清,如果其中含有切片中某种抗原的抗体,抗体便沉淀结合在抗原上,再用间接荧光抗体(抗种属特异性IgG荧光抗体)与结合在抗原上的抗体反应(如检测人血清中的抗体必需用抗人IgG荧光抗体等),在荧光显微镜下可见抗原抗体反应部位呈现明亮的特异性荧光。此法是检验血清中自身抗体和多种病原体抗体的重要手段(图3-3)
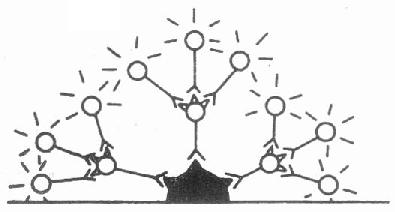
图3-3 间接法
3.检查抗原法双薄片此法是直接法的重要改进,先用特异性(对细胞或组织内抗原)抗体(或称第一抗体)与细胞标本反应,随后用缓冲盐水洗去未与抗原结合的抗体,再用间接荧光抗体(也称第二抗体,种特异性)与结合在抗原上的抗体(是第二抗体的抗原)结合,形成抗原-抗体-荧光抗体的复合物。由于结合在抗原抗体复合物上的荧光抗全显著多于直接法,从而提高了敏感性。如细胞抗原上每个分子结合3~5个分子抗体,当此抗体作为抗原时又可结合3~5分子的荧光抗体,所以和直接法相比荧光亮度可增强3至4倍。此法除灵敏性高外,它只需要制备一种种属间接荧光抗体,可以适用于多种第一抗体的标记显示。这是现在最广泛应用的技术。
三、补体法
1.直接检查组织内免疫复合物法用抗补体C3等荧光抗体直接作用组织切片,与其中结合在抗原抗体复合物上的补体反应,而形成抗原抗体补体复合物---抗补体荧光抗体复合物,在荧光显微镜下呈现阳性荧光的部位就是免疫复合物的存在处,此法常用于肾穿刺组织活检诊断等。
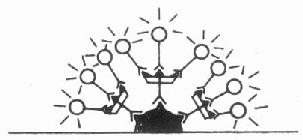
图3-4 补体法
2.间接检查组织内抗原法常将新鲜补体与第一抗体混合同时加在抗原标本切片上,经37℃孵育后,如发生抗原补体抗体反应,补体就结合在此复合物上,再用抗补体荧光抗体与结合的补体反应,形成抗原抗体—抗补体荧光抗体的复合物,此法优点是只需一种荧光抗体可适用于各种不同种属来源的第一抗体的标记显示。
四、双重免疫荧光标记法
在同一细胞组织标本上需要同时检查两种抗原时,要进行双重荧光染色,一般均采用直接法,将两种荧光抗体(如抗A和抗B)以适当比例混合,加在标本上孵育后,按直接法洗去未结合的荧光抗体,抗A抗体用异硫氰酸荧光素标记,发黄绿色荧光;抗B抗体用TMRITC或RB200标记,发红色荧光,可以明确显示两种抗原的定位。
五、对照试验
为了保证免疫荧光细胞化学染色的准确性,排除某些非特异性染色,必须在初次试验时进行以上对照试验:
1.直接法需设下述对照试验
(1)标本自发荧光对照:标本只加PBS或不加PBS,缓冲甘油封片,荧光显微镜观察应呈阴性荧光(无与特异性荧光相似的荧光)。
(2)抑制试验:可分为二步法和一步法。
①二步抑制法:标本先加未标记的特异性抗体,再加标记荧光抗体,结果应呈阴性或明显减弱的荧光。
②一步抑制法:先将荧光抗体用未标记抗体作适量混合,再加在标本上染色,结果应为阴性。此法效果较二步法好,并且简便。
(3)阳性对照:用已知阳性标本作直接法免疫荧光染色,结果应呈阳性荧光。
如对照(1)和(2)无荧光或弱荧光,(3)和待检查标本呈强荧光即为特异性阳性荧光。
2.间接法
(1)自发荧光对照:同上(一)。
(2)荧光抗体对照:标本只加间接荧光抗体染色,结果阴性。
(3)抑制试验:同上。
(4)阳性对照:同上。
如对照(1)、(2)、(3)均呈阴性,阳性对照和待检标本阳性则为特异性荧光。
3.补体法
(1)自发荧光对照
(2)荧光抗体对照
(3)抑制试验
(4)补体对照:取新鲜豚鼠血清1:10稀释先作用标本,再用抗补体荧光抗体染色,结果阴性。
(5)抑制试验:标本加灭活的第一抗体,再用1:10稀释度的新鲜豚鼠血清孵育后,再加未标记的抗补体血清与抗补体荧光抗体等量混合稀释液,结果应为阴性。
(6)阳性对照:(1)~(5)结果阴性,(6)和待检标本阳性时,则为特异性荧光。
第二节 荧光抗体的制备
荧光抗体是免疫荧光细胞化学的重要技术之一,制备高特异性和高效价的荧光抗体必须选用高质量的荧光素和高特异性高效价的免疫血清。
一、荧光素
荧光是指一个分子或原子吸收了给予的能量后即刻引起发光,停止能量供给,发光也瞬时停止(一般持续10-7~10-8s)。可以产生明亮荧光的染料物质,称荧光色素。目前主要常用的荧光色素有以下3种:
(一)异硫氰酸荧光素(Fluoresceinisothiocyanate, GITC)
呈黄色、橙黄或褐黄色粉末或结晶,性质稳定,在室温下能保存2年以上,在低温中可保存多年。易溶于水和酒精。最大吸收光谱为490~495nm,最大发射光谱为520~530nm,呈现黄绿色荧光,分子量为398.4(图3-5)。
在碱性条件下,FITC的异硫氰酸基在水溶液中与免疫球蛋白的自由氨基经碳酰胺化而形成硫碳氨基键,成为标记荧光免疫球蛋白,即荧光抗体。反应式如下(图3-6):
一个Ig分子上最多能 标记15~20个FITC分子。
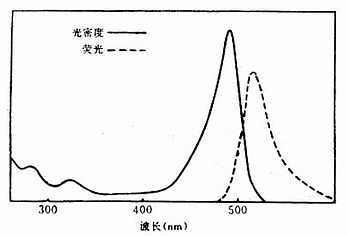
图3-5 FITC的吸收光谱和发射光谱
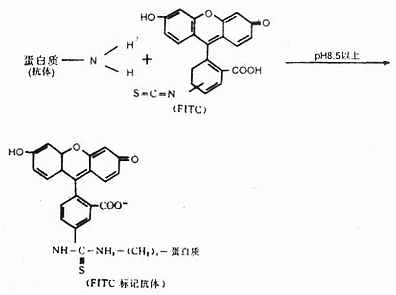
图3-6 抗体的FITC标记反应式
(二)四乙基罗达明(tetraethylrodamineB200, RB200)
呈褐红色粉末,不溶于水,易溶于酒精和丙酮,性质稳定,可长期保存。最大吸收光谱为570nm,最大发射光谱为595~600nm,呈明亮橙红色荧光。分子量为580(图3-7)。
RB200在五氯化磷(PCl5)作用下转变成磺酰氯(SO2Cl),在碱性条件下易与蛋白质的赖氨酸ε-氨基反应结合而标记在蛋白分子上。化学反应式如下(图3-8)。
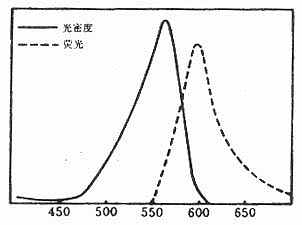
图3-7 RB200在可见光区的吸收
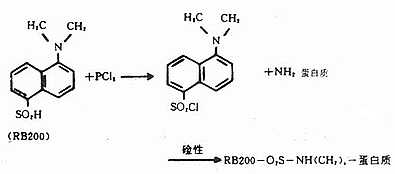
图3-8 RB200标记抗体反应光谱和荧光光谱
(三)四甲基异硫氰酸罗达明(tetramethylrhodamineisothiocyanate, TMRITC)
它是一种紫红色粉末,较稳定。其最大吸收光谱为550nm,最大发射光谱620nm,呈橙红色荧光,与FITC的黄绿色荧光对比清晰(图3-9),与蛋白质结合方式同TITC。它可用于双标记示踪研究。化学结构式如下(图3-10)。
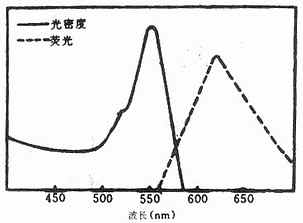
图3-9 TMRITC在可见光区的吸收光谱和发射光谱
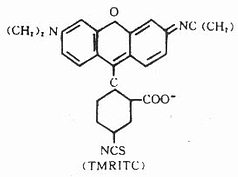
图3-10 TMRITC结构式
二、荧光素标记抗体的方法
(一)FITC标记抗体的方法
1.Marsshall 氏法
(1)材料:抗体球蛋白溶液、0.5mol/l pH9.0碳酸盐缓冲液、无菌生理盐水、异硫氰酸荧光素、1%硫柳汞水溶液、三角烧瓶(25~50ml)、冰及冰槽(或1000ml烧杯)、电磁搅拌器、灭菌吸管、透析袋、玻棒、棉线及烧杯(500ml)、pH7.2或8.0的0.01mol/L PBS等。
(2)方法及步骤:
①抗体的准备:取适量已知球蛋白浓度之溶液,置入三角烧瓶中,加入生理盐水及碳酸盐缓冲液,使最后蛋白浓度为20mg/ml,缓冲液容量为总量的1/10,混匀,将三角烧瓶置冰槽中,电磁搅拌(速度适当以不起泡沫为宜)5~10min。
②荧光素的准备:根据欲标记的蛋白质总量,按每毫克蛋白加0.01mg 荧光素,用分析天平准确称所取所需的异硫氰酸荧光素粉末。
③结合(或称标记):边搅拌边将称取的荧光色素渐渐加入球蛋白溶液中,避免将荧光素粘于三角烧瓶壁或搅拌玻棒上(大约5~10min内加完),加毕后,继续搅拌12~18h。结合期间应保持蛋白溶液于4℃左右,故须及时添冰去水;亦可将结合装置安放在4℃冰箱或冰库中。
④透析:结合完毕后,将球蛋白的溶液离心(2500r/min)20min,除去其中少量之沉淀物,装入透析袋中后再置于烧杯中,用pH8.0缓冲盐水透析(0~4℃)过夜。
⑤过柱:取透析过夜的标记物,过葡聚糖凝胶G-25或G-50柱,分离游离荧光素,收集标记的荧光抗体进行鉴定(图3-11,图-3-12)。
图3-11 Sephadex G-25 对FITC
图3-12 FITC与家兔IgG球蛋白在25℃和2℃时结合的动力学(Kawamura 1964)
洗脱液:0.01mol/L磷酸盐缓冲液(pH7.2);过滤量:12ml标记全球蛋白液(过滤前未透析);收集量:20ml(稀释1.7倍)。
分别以1mgFITC溶于2份1mol 0.5mol/L碳酸重碳酸盐缓冲液(分别为pH9.5和pH9.0),于2℃下搅拌将其各加入100mg家兔IgG生理盐水溶液中,搅匀后立即将每份分为两半。一半保留于2℃下,另一半置25℃下。间隔一定时间后各取出0.5ml通过sephadex G-25去游离FITC,由上计算出5mg家兔IgG结合的FITC量。
2.Chadwick 氏法
(1)材料:抗原球蛋白溶液、异硫氰酸荧光素、3%重碳酸钠水溶液、0.01mol/l Ph8.0磷酸盐缓冲盐水、1%硫柳汞、离心机及离心管、三角烧瓶(25ml)、冰槽、无菌吸管及毛细滴管、烧杯(500ml)、透析袋、棉线、玻棒等。
(2)方法及步骤:
①抗体准备:用0~4℃pH8.0磷酸盐缓冲盐水将球蛋白溶液稀释至浓度为30~40mg/ml,置入三角烧瓶内,放于冰槽中。
②荧光色素准备:按每毫克蛋白加入荧光素0.01mg计算,称取所需之荧光素量,用3%重碳酸钠水溶液溶解。
③将准备之抗体与荧光色素溶液等量混合,充分搅匀,在0~4℃,冰箱中结合18~24h。
④透析和柱层析:方法同Marshall 氏法。
3.改良法(1963年) 根据Marshall 氏法取高价的抗人球蛋白兔免疫血清,分离球蛋白,用盐水(0.15mol/l NaCl)及缓冲液(0.15mol/l NaHCO3 –Na2CO3 PH9.0) 稀释使每毫升内含蛋白10mg,缓冲液为总量的10%,降温至4℃,加入异硫氰酸盐荧光素,(蛋白:荧光素=50~80mg:1mg),在0~4℃下电磁搅拌12~14h。然后用半饱和和硫酸铵将标记球蛋白沉淀分离,除去未结合的荧光素,再用缓冲盐水透析,除去硫酸铵(用Nessler氏试剂测验至隔夜透析之盐水无氨离子及荧光色素为止)。将制备好的荧光抗体加叠氮钠0.01%,分装在1ml安瓿中,或冻干,保存于冰箱中(4℃)可以用半年以上,-20℃保存可达2年以上。
【附一】0.01mol/L pH7.2 PBS 配法:NaCl 18g 、Na2HPO41.5g、KH2PO40.2g,溶于2000ml蒸馏水中,校定pH至7.2。
【附二】0.5mol/L pH9.0碳酸盐缓冲液配法:取0.5mol/l Na2CO3(5.3%)10ml加入0.5mol/l NaHCO3(4.2%)90ml,混合后,校定pH至9.0。
【附三】3%重碳酸钠水溶液配法:称1.5g无水重碳酸钠充分溶解于50ml灭菌蒸馏水中即成。
4.透析标记法 此法适用于小量抗体的荧光素标记,标记简便,非特异性染色较少。
(1)用0.025mol/L碳酸盐缓冲液pH9.0,将欲标记免疫球蛋白稀释成1%浓度,装入透析袋中。
(2)用同一缓冲液将FITC配成0.1mg/ml的溶液,按1%球蛋白液体积的10倍,将FITC稀释液盛于圆柱形容器内,并使透析袋浸没于FITC液中。容器顶端盖紧,底部放搅拌棒,在4℃电磁搅拌下,透析标记24h。取出透析袋中标记液,即刻用sephadex G-50 凝胶过滤,去除游离荧光素,分装、贮存于4℃中(图3-13)。
图3-13 标记抗体溶液通过sephadex 产胶柱层析分布
(二)四乙基罗达明标记抗体方法
取1g RB200及PCL52g放在乳钵中研磨5min(在通风橱中)。然后加入10ml无水丙酮,放置5min,不断搅拌。过滤,用滤液进行标记抗体。剩余部分吸附在滤纸上,4℃干燥保存。
取抗体(20mg/ml)每毫升加入生理盐水和0.5mol/l pH9.0的碳酸盐缓冲液各1ml稀释。逐滴加入0.1ml RB200溶液,边加入边搅拌,在0~4℃中结合12~18h,再用生理盐水透析5~7h,经葡聚糖凝胶G-50柱层析,除去游离荧光素,分装,贮存于4℃备用。
(三)四甲基异硫氰酸罗达明标记抗体方法
(1)Igg 10ml (6mg/ml)在0.01mol/l pH9.5碳酸盐缓冲液中透析过夜。
(2)将四甲基异硫近氰酸罗达明(每毫克IgG加入5~20μg)溶于二甲亚砜(1mg/ml),取此溶液300μl,一滴一滴加入蛋白质溶液中,同时电磁搅拌。
(3)在室温中搅拌2h,避光。
(4)把结合物移入直径3cm,高30cm大小的Bio –Gel P-6层析柱(用0.01mol/l pH8.0的PBS平衡过),流速为1.5ml/min。
(5)收集先流出的红色结合物,即为标记抗体,分装,4℃保存备用。
(四)藻红蛋白标记抗体的方法
1.巯基化藻红蛋白(phycoerthrin PE)的制备,600μl的15.5mg/ml盐酸巯醇亚胺(iminothiolane hydrochloride)加到1.2ml的3.6mg/ml的PE中,和1.2ml PB、pH6.8 混合,装入透析袋置入50mmol/l pH6.8 PB中透析,4℃过夜,再换用pH7.5PB透析6h。每个PE分子中可结合8个巯基。
2.PE-IgG制备异双功能试剂SPDP[n - Succ - inimdyl3-(2-pyridyldlthio) propinate ] 30μl (1.1mg/ml)的乙醇溶液,加入700μl的4.2mg/ml IgG PB溶液(50mmol/l pH7.5),在室温中反应2.5h。再加入巯基化Pe 400μl(1.7mg/ml)加到500μl反应混合液中,室温反应12h,加入100μl的50mmol/L碘乙酸钠封闭残余巯基,用PB透析过夜,4℃。加入0.01%Na3N3分装,4℃保存半年。
(五)PE-标记蛋白A方法
(1)取4.08mg PE溶于0.1mol/l pH7.4PB(含0.1mol/l NaCl)1ml中,溶解后,取出0.5ml,再加入10μlSPDP无水甲醇液(2.65mg/ml),SPDP/蛋白摩尔比为10,22℃反应5min,过Sephadex G-50(1×17cm),用100mmol/l pH7.4 PBS(含0.1mol/l NaCl)平衡和洗脱。
(2)0.5ml蛋白(2mg/ml)100mmol/l PB(含有100mmol/l NaCl) pH7.4,加入2.6μl上述SPDP甲醇液,SPDP:蛋白=9.5,22℃,40min ,加入25μmol/L二硫苏糖醇(DTT)pH7.4缓冲液,22℃,25min,同上过Sephadex G-50,收集蛋白A峰。
(3)取0.77mg/ml的PE和0.27mg/ml蛋白A等量混合,22℃反应6h,混合物4℃保存备用。
以上两种PE标记制品,可最后溶于0.01mol/l pH7.4PB(含有0.1mol/l DETA、1mol/L碘乙酰胺、1%BSA 和0.1%NaN3),0~5℃保存。
(六)蓝色荧光素标记抗体方法
Kbaffan等(1986)首先创立了蓝色荧光素标记和染色技术,可进行双标记或多标记。
(1)取7-氨基-4-甲基香豆素(7-amino-4-methyl coumarin, AMC)260μg溶于二甲亚砜25μl中。
(2)将上液加入10ml IgG的 巴比妥缓冲液(0.5mol/L,pH8.5,内含50~100mg IgG)中,室温反应2h,过Sephadex G-50除去游离荧光素。
AMC呈黄色结晶固体,最大吸收波长354nm,最大荧光波长430nm。
三、荧光抗体质量控制
对制备的荧光抗体必须进行质量鉴定,主要进行特异性和敏感性两个方面的鉴定。
(一)染色特异性和敏感性的测定
1.特异性染色效价的测定直接染色以倍比稀释荧光抗体溶液如1:2,1:4,1:8……,与相应抗原标本作一系列染色,荧光强度在“+++”的最大释释度,即为该荧光抗体的染色滴度(效价)或单位。实际染色应用时,可取低一个或两个稀释度(即2~4个单位),如染色效价为1:64,实际应用时可取1:32或1:16。间接染色效价可按抗核抗体荧光染色法步骤,先用不同稀释度的荧光抗体染色,结果以抗核抗体荧光强度“++”为标准,染色用效价和直接法相同。
2.非特异性染色测定根据荧光抗体的用途不同,可用相类似的抗原切片或涂片,倍比稀释荧光抗体,按常规染色,结果在标本上出现的非特异染色应显著低于特异染色滴度,否则应采取消除非特异性染色的方法处理荧光抗体。
3.吸收试验在荧光抗体中加入过量相应抗原,于室温中搅拌2h后,移入4℃中过夜,3000r/min,离心30min,收集上清液,再用以染相应抗原阳性标本,结果应不出现明显阳性荧光。
4.抑制试验如前述。
(二)F/P比值的测定
F(荧光素)和P(抗体蛋白)的克分子比值反映荧光抗体的特异性染色质量,一般要求F/P的克分子比值为1~2。过高时,非特异性染色增强;过低时,荧光很弱,降低敏感性。
1.蛋白质定量 测定荧光抗体的蛋白质mg/ml量。
2.结合荧光素定量 先制作荧光素定量标准曲线,即准确称取FITC1mg,溶于10ml 0.5mol/L pH9.0碳酸盐缓冲液中,再用0.01mol/l pH7.2PBS稀释到100ml,此时荧光素含量为10 μg/ml,以此为原液,再倍比稀释9个不同浓度的溶液,用分光亮度计在490nm波长测定光密度值(OD),以光密度为纵坐标,荧光素含量为横坐标,作标准函数图。
荧光素与蛋白质结合后,其吸收光谱峰值向长波方向位移约5nm,FITC和蛋白质结合后由490nm变为493~495nm,RB200和蛋白质结合后变为595nm。
F/P比值的计算:可按以下公式计算。
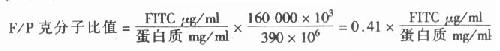
式中160000为抗体蛋白质的分子量,390为FITC的分子量。蛋白质从克换算为毫克需再乘以103,而荧光素从克换算为微克需要再乘以106。
测定RB200荧光抗体的克分子比值公式如下:

按图3-4测定法更为简便,即先用276nm波长测得蛋白质的OD值,再用493波长测得FITC的OD值,将这两个OD值在图3-14上连成一直线,直线与各纵线交叉处,即可查出标记抗体的以下数值:FITc μg/ml ,F/P 的μg/mg,F/P的克分子比值,蛋白mg/ml等。
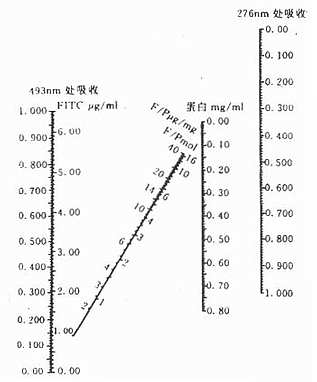
图3-14 FITC标记物中球蛋白、荧光色素和E/P比值计算图
四、荧光抗体的保存
以0~4℃或-20℃低温保存,防止抗体活性降低和蛋白变性。最好加入浓度为1:5000~10000的硫柳汞或1:1000~5000的叠氮钠防腐,小量分装如0.1~1ml,真空干燥后更易长期保存。
第三节 免疫荧光细胞化学染色方法
一、标本制作
可制作涂片、印片、细胞单层培养物、组织切片,经适当固定或不固定,作免疫荧光染色用。
二、荧光抗体染色方法
(一)直接法
1.染色 切片经固定后,滴加经稀释至染色效价如1:8或1:16的荧光抗体(如兔抗人γ-球蛋白荧光抗体或兔抗人IgG或IgA荧光抗体等),在室温或37℃染色30min,切片置入能保持潮湿的染色盒内,防止干燥。
2.洗片 倾去存留的荧光抗体,将切片浸入pH7.4或pH7.2PBS中洗两次,搅拌,每次5min,再用蒸馏水洗1min,除去盐结晶。
3.用50%缓冲(0.5mol/L碳酸盐缓冲液pH9.0~9.5)甘油封固、镜检
4.对照染色
①正常免荧光血清染色,如上法处理切片,结果应为阴性。②染色抑制试验(一步法):将荧光抗体和未标记的抗体球蛋白或血清(相同)等量混合,如上法处理切片。结果应为阴性。为证明此种染色抑制不是由于荧光抗体被稀释所致,可用盐水代替未标记抗血清,染色结果应为阳性。此法结果较二步法稳定。③类属抗原染色试验,前面已作叙述。
直接法比较简单,适合做细菌、螺旋体、原虫、真菌及浓度较高的蛋白质抗原如肾、皮肤的检查和研究。此法每种荧光抗体只能检查一种相应的抗原,特异性高而敏感性较低。
(二)间接法
(1)切片固定后用毛细滴管吸取经适当稀释的免疫血清滴加在其上,置于染色盒中保持一定的湿度,37℃作用30min。然后用0.01mol/l pH7.2PBS洗两次,10min,用吸水吸去或吹干余留的液体。
(2)再滴加间接荧光抗体(如兔抗人γ-球蛋白荧光抗体等),同上步骤,染色30min,37℃,缓冲盐水洗两次10min,搅拌,缓冲甘油封固,镜检。
对照染色:①抗体对照:用正常兔血清或人血清代替免疫血清,再用上法进行染色,结果应为阴性。②抗原对照:即类属抗原染色,亦应为阴性。③阳性对照。
间接法中上述方法称双层法(Double Layer Method)。另一种称夹心法,即用未标记的特异性抗原加在切片上先与组织中之相应抗体结合,再用该抗原之荧光抗体重叠结合其上,而间接地显示出组织和细胞中抗体的存在,方法步骤如下:
①切片或涂片固定后,置于染色湿盒内。
②滴加未标记的特异性抗原作用切片于37℃,30min。
③缓冲盐水洗2次,每次5min,吹干。
④滴加特异性荧光抗体再用切片于37℃,30min。
⑤如③水洗。
⑥缓冲甘油封固,镜检。
间接法只需制备一种荧光抗体可以检出多种抗原,敏感性较高,操作方法较易掌握,而且能解决一些不易制备动物免疫血清的病原体(如麻疹)等的研究和检查,所以已被广泛应用于自身抗体和感染病人血清的试验。
(三)补体法
1.材料
(1)免疫血清60℃灭活20min,用Kolmers 盐水作2倍稀释成1:2,1:4,1:8……。补体用1:10稀释的新鲜豚鼠血清,抗补体荧光抗体等,按下述的补体法染色。免疫血清补体结合的效价,如为1:32则免疫血清应作1:8稀释。
(2)补体用新鲜豚鼠血清一般作1:10稀释或按补体结合反应试管法所测定的结果,按2单位的比例,用Kolmers盐水稀释备用。Kolmers盐水配法:即在pH7.4、0.1mol/L的磷酸缓冲盐水中,溶解MgSO4的含量为0.01%浓度。
(3)抗补体荧光抗体:在免疫血清效价为1:4,补体为2单位的条件下,用补体染色法测定免疫豚鼠球蛋白荧光抗体的染色效价,然后按染色效价1:4的浓度用Kolmers盐水稀释备用。
2.方法步骤
(1)涂片或切片固定。
(2)吸取经适当稀释之免疫血清及补体之等量混合液(此时免疫血清及补体又都稀释一倍)滴于切片上,37℃作用30min,置于保持一定湿度的染色盒内。
(3)用缓冲盐水洗2次,搅拌,每次5min,吸干标本周围水液。
(4)滴加经过适当稀释之抗补体荧光抗体30min,37℃,水洗同(3)。
(5)蒸馏水洗1min,缓冲甘油封固。
3.对照染色
(1)抗原对照。
(2)抗血清对照:用正常兔血清代替免疫血清。
(3)灭活补体对照:将补体经56℃30min处理后,按补体同样比例稀释,与免疫血清等量混合后,进行补体法染色。
本法之荧光抗体不受免疫血清的动物种属的限制,因而一种荧光抗体可作更广泛的应用,敏感性亦较间接法高,效价低的免疫血清亦可应用,节 省免疫血清,尤其是对检查形态小的如立克氏体、病毒颗粒等或浓度较低的抗原物质时甚为理想。
(四)膜抗原荧光抗体染色法
本法应用直接法或间接法的原理和步骤,可对活细胞在试管内进行染色,常用于T和B细胞、细胞培养物、瘤细胞抗原和受体等的检查和研究,阳性荧光主要在细胞膜上。FACS即采用此法原理。
(五)双重染色法
在同一标本上有两个抗原需要同时显示(如A抗原和B抗原),A抗原的抗体用FITC标记,B抗原的抗体用罗达明标记,可采用以下染色方法:
1.一步法双染色 先将两种标记抗体按适当比例混合(A+B),按直接法进行染色。
2.二步法双染色 先用RB200标记的B抗体染色,不必洗去,再用FITC标记的A抗体染色,按间接法进行。
结果:A抗原阳性荧光呈现绿色,B抗原阳性呈现桔红色荧光。
(六)荧光抗体再染色法
若切片或其他标本经某种荧光抗体染色后,未获得阳性结果,而又疑有另外的病原体存在时,可用相应的荧光抗体再染色。
有时存档蜡块不能再用以切片,也可用存档的HE染色标本,褪去盖片和颜色,再作免疫荧光或其它免疫细胞化学的染色。
三、荧光抗原染色法
某些抗原可以用荧光素标记,制成荧光抗原,标记荧光素的方法与制备荧光抗体方法相同。用荧光抗原可以直接检查细胞或组织内的相应抗体,特异性较好,敏感性较差。染色方法同荧光抗体染色的直接法。由于多数抗原难以提纯或量少不昂贵,一般很少采用此法。
第四节 荧光显微镜检查法
一、荧光显微镜
荧光显微镜是免疫荧光细胞化学的基本工具。它是由光源、滤板系统和光学系统等主要部件组成。是利用一定波长的光激发标本发射荧光,通过物镜和目镜系统放大以观察标本的荧光图像(图3-15)。
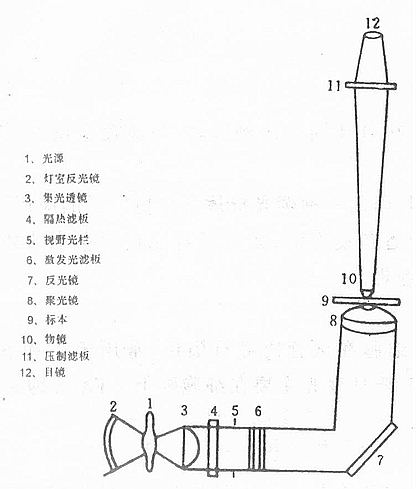
图3-15 荧光显微镜的结构和主要部件
(一)光源
现在多采用200W的超高压汞灯作光源,它是用石英玻璃制作,中间呈球形,内充一定数量的汞,工作时由两个电极间放电,引起水银蒸发,球内气压迅速升高,当水银完全蒸发时,可达50~70个标准大气压力,这一过程一般约需5~15min。超高压汞灯的发光是电极间放电使水银分子不断解离和还原过程中发射光量子的结果。它发射很强的紫外和蓝紫光,足以激发各类荧光物质,因此,为荧光显微镜普遍采用。
超高压汞灯也散发大量热能。因此,灯室必须有良好的散热条件,工作环境温度不宜太高。
新型超高压汞灯在使用初期不需高电压即可引燃,使用一些时间后,则需要高压启动(约为15000V),启动后,维持工作电压一般为50~60V,工作电流约4A左右。200W超高压汞灯的平均寿命,在每次使用2h的情况下约为200h,开动一次工作时间愈短,则寿命愈短,如开一次只工作20min,则寿命降低50%。因此,使用时尽量减少启动次数。灯泡在使用过程中,其光效是逐渐降低的。灯熄灭后要等待冷却才能重新启动。点燃灯泡后不可立即关闭,以免水银蒸发不完全而损坏电极,一般需要等15min。由于超高压汞灯压力很高,紫外线强烈,因此灯泡必须置灯室中方可点燃,以免伤害眼睛和发生爆炸时造成操作。
超高压汞灯(100W或200W)光源的电路和包括变压、镇流、启动几个部分。在灯室上有调节 灯泡发光中心的系统,灯泡球部后面安装有镀铝的凹面反射镜,前面安装有集光透镜。
国产超高压汞灯GCQ-200型性能良好,可以代替HBO-200等型的进口灯泡,平均寿命在200h以上,价格也比较低。
我国研制的一种简易轻便型高色温溴钨荧光光源装置,体积小,重量轻,功率小,交、直流两用(自带直流电源),易于携带,使用方便,已推广应用。
(二)滤色系统
滤色系统是荧光显微镜的重要部位,由激发滤板和压制滤板组成。滤板型号,各厂家名称常不统一。滤板一般都以基本色调命名,前面字母代表色调,后面字母代表玻璃,数字代表型号特点。如德国产品(Schott)BG12,就是种蓝色玻璃,B是蓝色的第一个字母,G是玻璃的第一个字母;我国产品的名称已统一用拼音字母表示,如相当于BG12的蓝色滤板名为QB24,Q是青色(蓝色)拼音的第一个字母,B是玻璃拼音的第一个字母。不过有的滤板也可以透光分界滤长命名,如K530,就是表示压制滤长530nm以下的光而透过530nm以上的光。还有的厂家的滤板完全以数字命名,如美国Corning厂的NO:5-58,即相当于BG12。
用于荧光显微镜的主要滤板如表3-1。
表3-1 荧光显微镜常用滤板型号和透光特点
| 基本色调 | 相应名称 | 2mm厚透光范围(峰值)nm | |||
| 上海电器元件厂 | 德国(Schott) | 苏联 | 日本 | ||
| 黑紫 | ZWB-1 | UG-1 | yΦC-2 | DV-1 | 300~400(365) |
| 黑紫 | ZWB-2 | UG-5 | yΦC-1 | 280~240(360) | |
| 靛蓝 | ZB-2 | BG-1 | ΦC-1 | BG-1 | 300~500(380) |
| 靛蓝 | ZB-3 | BG-3 | CC-4 | BG-3 | 260~520(400) |
| 靛蓝 | QB-24 | BG-12 | CC-8 | BG-12 | 310~570(420) |
| 淡蓝 | QB-10 | BG-38 | C3C-5 | 310~720(460) | |
| QB-12 | -8 | ||||
| -11 | |||||
| 橙黄 | CB-3 | OG-1(K530) | OC-11 | OG-1 | 530以上 |
| 橙黄 | JB-8 | OG-4(K510) | ЖC-18 | FY-5 | 510以上 |
| 绿橙 | JB-7 | GG-11(K490) | ЖC-17 | FY-3 | 480以上 |
| FY-4 | |||||
| 淡绿 | JB-4 | GG-3(K430) | жC-11 | US-10 | 420以上 |
引自:五九一节 五部队编:荧光显微术,见参考资料)
1.激发滤板 根据光源和荧光色素的特点,可选用以下三类激发滤板,提供一定波长范围的激发光。
紫外光激发滤板:此滤板可使400nm以下的紫外光透过,阻挡400nm以上的可见光通过。常用型号为UG-1或UG-5,外加一块BG-38,以除去红色尾波。
紫外蓝光激发滤板:此滤板可使300~450nm范围内的光通过。常用型号为ZB-2或ZB-3,外加BG-38。
紫蓝光激发滤板:它可使350~490nm的光通过。常用型号为QB24(BG12)。
最大吸收峰在500nm以上者的荧光素(如罗达明色素)可用蓝绿滤板(如B-7)激发。
近年开始采用金属膜干涉滤板,由于针对性强,波长适当,因而激发效果比较玻璃滤更好。如西德Leitz厂的FITC专用KP490滤板和罗达明的S546绿色滤板,均远比玻璃滤板效果好。
激发滤板分薄厚两种,一般暗视野选用薄滤板,亮视野荧光显微镜可选用厚一些。基本要求是以获得最明亮的荧光和最好的背景为准。
2.压制滤板 压制滤板的作用是完全阻挡激发光通过,提供相应滤长范围的荧光。与激发滤板相对应,常用以下3种压制滤板:
紫外光压制滤板:可通过可见光、阻挡紫外光通过。能与UG-1或UG-5组合。常用GG-3K430或GG-6K460。
紫蓝光压制滤板:能通过510nm以上滤长的荧光(绿到红),能与BG-12组合。通常用OG-4K510或OG-1K530。
紫外紫光压制滤板:能通过460nm以上波长的荧光(蓝到红),可与BG-3组合,常用OG-11K470AK 490,K510。
(三)反光镜
反光镜的反光层一般是镀铝的,因为铝对紫外光和可见光的蓝紫区吸收少,反射达90%以上,而银的反射只有70%;一般使用平面反光镜。
(四)聚光镜
专为荧光显微镜设计制作的聚光器是用石英玻璃或其他透紫外光的玻璃制成。分明视野聚光器的暗视野聚光器两种。还有相差荧光聚光器。
1.明视野聚光器 在一般荧光显微镜上多用明视野聚光器,它具有聚光力强,使用方便,特别适于低、中倍放大的标本观察。
2.暗视野聚光器 暗视野聚光器在荧光显微镜中的应用日益广泛。因为激发光不直接进入物镜,因而除散射光外,激发光也不进入目镜,可以使用薄的激发滤板,增强激发光的强度,压制滤板也可以很薄,因紫外光激发时,可用无色滤板(不透过紫外)而仍然产生黑暗的背景。从而增强了荧光图像的亮度和反衬度,提高了图像的质量,观察舒适,可能发现亮视野难以分辨的细微荧光颗粒。
3.相差荧光聚光器 相差聚光器与相差物镜配合使用,可同时进行相差和荧光联合观察,既能看到荧光图像,又能看到相差图像,有助于荧光的定位准确。一般荧光观察很少需要这种聚光器。
(五)物镜
各种物镜均可应用,但最好用消色差的物镜,因其自体荧光极微且透光性能(波长范围)适合于荧光。由于图像在显微镜视野中的荧光亮度与物镜镜口率的平方成正比,而与其放大倍数成反比,所以为了提高荧光图像的亮度,应使用镜口率大的物镜。尤其在高倍放大时其影响非常明显。因此对荧光不够强的标本,应使用镜口率大的物镜,配合以尽可能低的目镜(4×,5×,6.3×等)。
(六)目镜
在荧光显微镜中多用低倍目镜,如5×和6.3×。过去多用单筒目镜,因为其亮度比双筒目镜高一倍以上,但目前研究型荧光显微镜多用双筒目镜,观察很方便。
(七)落射光装置
新型的落射光装置是从光源来的光射到干涉分光滤镜后,波长短的部分(紫外和紫蓝)由于滤镜上镀膜的性质而反射,当滤镜对向光源呈45。倾斜时,则垂直射向物镜,经物镜射向标本,使标本受到激发,这时物镜直接起聚光器的作用。同时,滤长长的部分(绿、黄、红等),对滤镜是可透的,因此,不向物镜方向反射,滤镜起了激发滤板作用,由于标本的荧光处在可见光长波区,可透过滤镜而到达目镜观察,荧光图像的亮度随着放大倍数增大而提高,在高放大时比透射光源强。它除具有透射式光源的功能外,更适用于不透明及半透明标本,如厚片、滤膜、菌落、组织培养标本等的直接观察。近年研制的新型荧光显微镜多采用落射光装置,称之为落射荧光显微镜。
二、荧光显微镜标本制作要求
(一)载玻片
载玻片厚度应在0.8~1.2mm之间,太厚的坡片,一方面光吸收多,另一方面不能使激发光在标本上聚集。载玻片必须光洁,厚度均匀,无明显自发荧光。有时需用石英玻璃载玻片。
(二)盖玻片
盖玻片厚度在0.17mm左右,光洁。为了加强激发光,也可用干涉盖玻片,这是一种特制的表面镀有若干层对不同波长的光起不同干涉作用的物质(如氟化镁)的盖玻片,它可以使荧光顺利通过,而反射激发光,这种反射的激发光女可激发标本。
(三)标本
组织切片或其他标本不能太厚,如太厚激发光大部分消耗在标本下部,而物镜直接观察到的上部不充分激发。另外,细胞重迭或杂质掩盖,影响判断。
(四)封裱剂
封裱剂常用甘油,必须无自发荧光,无色透明,荧光的亮度在pH8.5~9.5时较亮,不易很快褪去。所以,常用甘油和0.5mol/l pH9.0~9.5的碳酸盐缓冲液的等量混合液作封裱剂。
(五)镜油
一般暗视野荧光显微镜和用油镜观察标本时,必须使用镜油,最好使用特制的无荧光镜油,也可用上述甘油代替,液体石蜡也可用,只是折光率较低,对图像质量略有影响。
三、使用荧光显微镜的注意事项
(1)严格按照荧光显微镜出厂说明书要求进行操作,不要随意改变程序。
(2)应在暗室中进行检查。进入暗室后,接上电源,点燃超高压汞灯5~15min,待光源发出强光稳定后,眼睛完全适应暗室,再开始观察标本。
(3)防止紫外线对眼睛的损害,在调整光源时应戴上防护眼镜。
(4)检查时间每次以1~2h为宜,超过90min,超高压汞灯发光强度逐渐下降,荧光减弱;标本受紫外线照射3~5min后,荧光也明显减弱;所以,最多不得超过2~3h。
(5)荧光显微镜光源寿命有限,标本应集中检查,以节 省时间,保护光源。天热时,应加电扇散热降温,新换灯泡应从开始就记录使用时间。灯熄灭后欲再用时,须待灯泡充分冷却后才能点燃。一天中应避免数次点燃光源。
(6)标本染色后立即观察,因时间久了荧光会逐渐减弱。若将标本放在聚乙烯塑料袋中4℃保存,可延缓荧光减弱时间,防止封裱剂蒸发。
(7)荧光亮度的判断标准:一般分为四级,即“一”—无或可见微弱荧光。“+”—仅能见明确可见的荧光。“++”—可见有明亮的荧光。“+++”—可见耀眼的荧光。
四、荧光图像的记录方法
荧光显微镜所看到的荧光图像,一是具有形态学特征,二是具有荧光的颜色和亮度,在判断结果时,必须将二者结合起来综合判断。结果记录根据主观指标,即凭工作者目力观察。作为一般定性观察,基本上可靠的。随着技术科学的发展,在不同程度上采用客观指标记录判断结果,如用细胞分光亮度计,图像分析仪等仪器。但这些仪器记录的结果,也必须结合主观的判断。
荧光显微镜摄影技术对于记录荧光图像十分必要,由于荧光很易褪色减弱,要即时摄影记录结果。方法与普通显微摄影技术基本相同。只是需要采用高速感光胶片如ASA200以上或24。以上。因紫外光对荧光猝灭作用大,如FITC的标记物,在紫外光下照射30s,荧光亮度降低50%。所以,曝光速度太慢,就不能将荧光图像拍摄下来。一般研究型荧光显微镜都有半自动或全自动显微摄影系统装置。
第五节 非特异性染色的消除方法
一、非特异性染色的主要因素
组织的非特异性染色的机理很复杂,其产生的原因主要可分为以下几点:
(1)一部分荧光素未与蛋白质结合,形成了聚合物和衍化物,而不能被透析除去。
(2)抗体以外的血清蛋白与荧光素结合形成荧光素脲蛋白,可与组织成分结合。
(3)除去检查的抗原以外,组织中还可能存在类属抗原(如Forssman氏抗原),可与组织中特异性抗原以外之之相应抗体结合。
(4)从组织中难于提纯抗原性物质,所以制备的免疫血清中往往混杂一些抗其他组织成分的抗体,以致容易混淆。
(5)抗体分子上标记的荧光素分子太多,这种过量标记的抗体分子带过多的阴离子,可吸附于正常组织上而呈现非特异性染色。
(6)荧光素不纯,标本固定不当等。
二、消除非特异性染色的方法
消除荧光抗体非特异性染色的方法应根据产生的原因采取适当的方法,常用的方法有以下几种:
(一)动物脏器粉末吸收法
常用肝粉(猪、大白鼠或小白鼠),其次是骨髓粉、鼠脑粉和鸡胚粉等。每毫升荧光抗体中加入肝粉50~100mg,在离心管中充分混匀,在室温中振动2h,4℃中过夜,再搅拌10min,高速离心(3000~15000r/min)30min,1~2次后,即可使用其上清液。吸收一般应在临用前进行,吸收后之荧光抗体保存冰箱中勿超过2周。染色应作吸收前后之比较,吸收时可先用缓冲盐水将组织干粉浸湿,离心(3000~15000r/min)30min,除去上清液,再加入荧光抗体进行吸收,以免消耗过多的抗体。
肝粉或新鲜细胞吸收是一种非特异性的消除方法,对荧光抗体的荧光色素和蛋白都有吸附作用。如检查组织中的病毒抗原时,也可用相同的组织干粉或匀浆沉淀物吸收之。
用脏器肝粉吸收对荧光抗体损失较多,如果根据Hiramotos氏等的方法将组织的20%生理盐水匀浆液,用生理盐水洗2~3次,12000r/min 10min离心沉淀,用其沉淀物吸收其荧光抗体即能完全达到目的,京极方久氏认为这样吸收对荧光抗体几乎没有损失,他们常用此法,效果甚佳,吸收后放置一周左右,用时有必要再吸收一次。
【肝粉的制法】
(1)将若干只小白鼠或大白鼠放血杀死,取出肝脏,用生理盐水洗2~3次,除去血液,剥掉表面的结缔组织的脂肪。
(2)剪碎,用生理盐水反复洗涤至无血色止,然后再加生理盐水少许,用组织捣碎机或匀浆器作成匀浆。
(3)将肝匀浆装入离心管内(1/3左右),交换地用2~3倍量生理盐水和丙酮反复洗涤各三次,至上清无血色止,每次完毕先用2000r/min离心沉淀15min后,再除去上清液。
(4)最后用丙酮洗涤肝浆,再用布氏漏斗过滤,或离心沉淀,将沉淀物平铺在洁净的玻璃板上,37℃烤干(过夜)。
(5)在乳钵中充分研磨,用120目铜筛筛选过后,分装,密封,低温干燥保存。
(二)透析法
荧光素如FITC分子可以通过半透膜,而蛋白质大分子不能透过,可将未与蛋白结合的荧光素透析除去。
(1)将标记完毕的荧光蛋白液装入一透析袋或玻璃纸袋内,液面稍留空隙,紧扎。
(2)浸入0.02mol/ph 7.1~7.4的PBS中(悬于大于标记物体积约50~100倍的PBS内),在4℃中透析,每日更换3~4次PBS,约5~7天,透析液中无荧即可(在荧光光源照射下)。
(三)葡聚糖凝胶G-50柱层析法
除游离荧光素可用2×46cm柱层析法,详细方法参阅第二章 。加入荧光抗体15~18ml(按床体积的5%~10%加样),使其缓慢渗入柱内,待即将全部入柱时,加入PBS少许,关闭下口,停留30~40min ,使游离荧光充分进入细筛孔中,然后再接通洗脱瓶开始滴入洗脱液。加入洗脱液一定量后,荧光抗体即向下移行,逐渐与存留于上端的游离荧光素之间拉开明显的界线,随着大量洗脱液的不断加入,二者分离距离越来越大,荧光抗体最先流出,分前、中、后三部分收集,测F/P比值,合格者合并,浓缩,分装。洗脱液用20%磺基水杨酸测定蛋白(发生沉淀反应),继续洗脱,游离荧光素则相继被洗脱下来,至洗脱液中无蛋白和荧光素后,此层析柱即可再用。
若用以除去荧光抗体中的游离荧光素和硫酸铵等盐类,可先在过柱前透析一夜,否则,NH4+太浓,在蛋白未完全洗脱时即出现NH4+,因而影响提纯与回收蛋白,一般待洗脱液出现蛋白时,即进行收集,之后出现SO4++(用1%BaCl2检查发生白色沉淀)。最后是NH4+,(用纳氏试剂检查呈黄棕色沉淀),待洗脱液无SO4++及NH4+后可再用。
如仅用小量荧光抗体,可用1×20cm的柱层析柱,取2g Sephadex G-50装柱,即可过滤2~3.5ml荧光抗体。
(四)DEAE纤维素柱层析法
标记过多或过少荧光素的抗体分子可用DEAE-纤维素柱层析法除去。方法如下:
DEAE-纤维素柱的装柱,洗脱、再生方法等与提纯IgG方法相同。装柱所需DEAE-纤维素量以干重每克交换20~50mg标记蛋白量为宜。常用梯度洗脱法如下:
(1)层析柱用0.01mol/L、pH7.2PB平衡,标记物上柱后,先用0.01mol/L、pH7.2PB洗脱,洗出无色或淡绿色液体,洗脱液量(根据床体积大小每梯度乘3),然后依下列各种离子强度洗脱液,分别洗脱和收集:
0.01mol/L、pH7.2PBS(0.05mol/l NaCl)……洗脱部分1。
0.01mol/L、pH7.2PBS(0.01mol/l NaCl)……洗脱部分2。
0.01mol/L、pH7.2PBS(0.02mol/l NaCl)……洗脱部分3。
将此三部分收集液(每管5ml)分别测定其F/P比值,0.05mol/l NaCl pH7.2PB洗脱液280nm光密度高峰管合并,浓缩保存备用。因这部分非特异性染色荧光最少,是比较好的荧光抗体。其他两部分可以废弃。
(2)柱上吸附的过度标记蛋白可继续增加NaCl的浓度至2.0mol/L洗脱完。
经过DEAE-纤维素层析后的标记抗体,其抗体量一般约损失50%,因此有些要求不太高的抗体,如抗细菌荧光抗体,不一定要这样处理,可用染色效价测定的稀释法除去非特异性染色。
(五)荧光抗体稀释法
先测定荧光抗体特异性染色与非特异性染色的效价,若二者效价相差较大,则可将荧光抗体稀释至一临界浓度,使特异性染色呈阳性,而使非特异性染色保持阴性,稀释方法和染色效价测定方法相同。
(六)纯化抗原法
用各种方法提纯单一成分的抗原是产生单价特异性抗体的最主要条件。近代免疫化学技术(免疫吸收法)和柱层析法等提供了很大的可能性,可参考有关专着。
(七)纯化抗体法---免疫吸收法
例如抗IgA血清的纯化方法---免疫吸收法。如分泌型IgA(SIgA)抗原纯度不高,所制的抗血清常与IgG呈交叉反应,为此需要吸收,常采用纯化的人IgG戊二醛聚合物加以吸收纯化。方法如下:
1.人IgG聚合物的制备在5ml含40mg/ml人IgG的0.1mol/l pH7.0磷酸缓冲溶液中,加入2.5%戊二醛溶液1ml,边加边搅,5min即出现混浊,逐现大块胶块,放置30min后,用研钵将凝胶磨细,继用1.0mol/l pH7.0磷酸缓冲溶液反复洗涤3次,末次加蒸馏水至20ml,即为人IgG聚合物悬液。
2.免疫吸收法将待吸收的抗SIgG血清加入待量IgG聚合物悬液,置室温搅拌60min,离心沉淀,上清液即稀释1倍的纯化抗SIgA血清。如用IgG聚合物作少量分次吸收,其效果更好。
(八)伊文氏蓝(Evans blue)衬染法
用0.01%伊文氏蓝的0.01mol/l pH7.2PBS稀释荧光抗体,可将背景细胞和组织染色,呈红色荧光,与特异性黄绿色荧光形成鲜明的对比,减少了非特异性荧光,宜作常规应用。伊文氏蓝一般先配成1%溶液,保存于4℃,用前再稀释至0.01%用以和然释荧光抗体。
此外,还可以用胰酶消化组织切片或用10%牛血清蛋白封闭法等消除非特异性染色,提高特异性染色。
参考文献
1.Coons AH, et al. Localization ofantigen in tissue cell.II. Improvements in a method for the detection ofantigen by means of fluorescent an-tibody. J Exp Med, 1950;91:1-3
2.RiggsJL, Seiwald RI, Burckhulter J, Powns CM and Metcalf TG. Iosthiocyanatecompounds as fluorescent labeling agents for immune serum. Am J Path,1958;34:1081~1097
3.MarshallID Jr, Eveland WC and Smith CW. Superiority of fluorescein isothiocyanate(Riggs)for fluorescent antibody technique with a modi-ification of its application. Procsec Exper Biol C Med(NP), 1958;98:898~900
4.GoldsteinG, Slizys IS and Chase MW. Stuides on fluoresent staining.1. Nonspecificfluorescence with fluorescein - coupled sheep anti-rab-bit globulins. J ExperMed, 1961;114:89~110
5.NairnRC. Fluorecent protein tracing. E 35:383
8.GotakoYamad, et al .Hepatitis B core and surface antigens in liver tissue. LabInvest, 1977;36(6):649
9.黄 策.免疫荧光和荧光免疫.1983年全军三防医学会议报告论文
10.张忠兵,等.双标记免疫荧光法对胃粘膜抗体产生细胞的对比研究.上海免疫学杂志,1983;3(5):304
11.FairbanksTR. Current status of lymphocyte subpopulation testing in humans. Am J MedTechnol, 1980;46:471
12.李元敏等译,免疫细胞化学.原子能出版社,1985:25~44
13.WickG, Traill KN, Schouestein K. Immunofluorescence Technology, SelectedTheoretical and Clinical Aspects. Elseries Biomedical Press. Amsterdom,1982:P27~36, P181, P219~262, P317~323
14.AKiyoshi Kawamura Jr. Fluorescent antibody . Technique and Their Applications.Second Edition University of Tokyo Press, Tokyo,1977:P10,P79,P141~281
15. RayMB, et al. Immunofluorescent detection of alphaantitrypsin in paraffin embeddedliver. J Clin Pathol, 1975;28:717
16.王伯沄,胰酶消化法提高甲醛固定石蜡切片免疫组织化学法敏感性方法,第四军医大学学报,1982;2:127
17. AurelF. Labelled antibodies in biology and medicine.Printed in Romania,1978:PP52-190
18. 王伯沄,免疫荧光技术:见汪美先 主编.免疫学基础.西安:陕西省科学技术出版社,1981:279-295
19. 59175部队.荧光显微术.上海科学技术情报所,1976:224~288
20. PolakJM and Noorden SV. Immunocytochemistry, PP233~248Wright, PSG. 1983
21李成文.现代免疫化学技术.上海:上海科技出版社,1992:97~100
第四章 免疫酶细胞化学
免疫酶细胞化学是免疫细胞化学(Immunocytochemistry,ICC)中最常用的方法之一,它是在抗原抗体特异反应存在的前提条件下,借助于酶细胞化学的手段,检测某种物质(抗原/抗体)在组织细胞内存在部位的一门新技术:即预先将抗体与酶连结,再使其与组织内特异抗原反应,经细胞化学染色后,于光镜或电子显微镜下观察分析的形态学研究方法。
40多年前,Coons及其同事利用荧光色素(FITC)标记抗体而开创的免疫荧光抗体技术,具有一定的灵敏性、特异性、操作简单等优点,但亦存在抗体用量大,标本不能长期保存、需较昂贵的荧光显微镜等问题。几乎在同一时期,电子显微镜在医学生物学领域中得到了广泛的应用。为克服荧光抗体法的不足、并能在超微结构水平定位抗原物质的存在部位,Nakane(1966)等成功地引入了酶代替荧光色素标记抗体,进行组织细胞内抗原或半抗原的定位,开辟了酶标抗体技术免疫酶细胞化学之路。
Sternberger(1970)等人在此基础上又作了改良,建立了非标记抗体酶法以及PAP法。酶标抗体法确立至今已经27个春秋,不断发展、成熟。各种新技术的引入,使新抗体相继问世,应运而生的抗体制造、经销商遍布世界各地,使ICC技术得到了更广泛的推广和应用,成为当今形态学研究领域中不可缺少的手段;同时也有为临床病理诊断、肿瘤性质的判定、预后的估测等提供重要依据,是临床实验室常规检查法之一。近年,伴随分子生物学、分子遗传学的惊人进步,ICC技术在基因表达产物的观察,细胞功能动态分析中,亦发挥着重要作用。在此,结合,笔者经验,介绍ICC染色中的主要程序:组织标本的固定、切片、染色原理及步骤、结果判定及一些进展、特殊应用等供读者参考。
第一节 固定和切片
一、固定
若想得到理想的ICC染色结果、正确地判断抗原物质在组织细胞内的位置,除需有良好酶和抗体外,保持组织细胞内抗原物质的不动性(Immobility)和免疫活性也是至关重要的。换言之,如果抗原物质在组织细胞间弥散、丢失或失去免疫活性,无论如何努力染色都是徒劳的,所以说固定是ICC染色中非常重要的一环。ICC与其它组织学技术不同,除要求保存良好的结构外,还需保存组织抗原的免疫活性。一般认为,新鲜组织能够最大限度地保存组织抗原的免疫活性,但结构较差,易出现抗原弥散丢失现象。Cumming(1980)报告,不固定的组织切片ICC染色时,环鸟苷酸含量丢失80%以上。固定的目的是使构成组织细胞成分的蛋白等物质不溶于水和有机溶剂,并迅速使组织细胞中各种酶降解、失活,防止组织自溶和抗原弥散,保持组织细胞的完整性和所要检测物质的抗原性。因此迅速充分固定是ICC染色成功的关键一步。用于ICC的固定剂种类较多,选择时应根据所要检测物质的抗原性质和切片方法以及所用抗体特征等做最佳筛选。制片及固定方法见第一章 ,下面介绍两种近年报道的新方法。
1.AMEX(Acetone Meth Enzoate Xylene )法是改良的冰冻置换法(freeze substitution)的简称,主要用于石蜡包埋标本。Sato(1986)报告,该法具有同新鲜(未固定)组织冰冻切片同样的抗原保持性和石蜡切片的良好组织结构保存。其机制为:组织在丙酮中固定(脱水),组织细胞内水份逐渐被丙酮取代,继之,用苯甲酸酯取代组织内丙酮,经二甲苯置换后,石蜡包埋。组织在-20℃丙酮内过夜亦形成冰晶,所以原方法将组织先置4℃丙酮20~30min,再移入-20℃丙酮过夜。实际上组织在4℃丙酮内过夜亦可得到同样效果。如在该固定液内加入少量蛋白酶活性阻断剂,并采用低溶点石蜡包埋等,可获得更佳染色结果。
2.微波固定(Microwave fixation)为近几年所注目的问题之一。该方法能保持良好的组织结构和抗原性,适于各种切片的酶组化、ICC以及免疫电镜等材料固定。其固定机理可能与微波(频率1000~3000MHz)具有被水分吸收的性质(通常所用的微波是频率2450MHz,波长12cm);生物材料含有大量水份,照射后,温度升高,分子运动加快,促进固定液向组织内渗透,加速与组织成分的反应,短时间内达到固定的效果等有关。经微波照射固定的组织,需置相同固定液中,继续固定2~6h,室温。
近年研究的专门固定用的微波炉已商品化,例如:Bio-Rad H2500型、日新EM·MWE-2等,该类微波炉能够准确地调节 照射时间和测定被照材料的瞬时温度。利用家庭微波炉代替时,应选用能以秒为单位调控的微波炉。实验材料不同,所需固定液的量、照射时间等各异,所以应在预实验的基础上,找出合适的固定液量、照射时间和温度。多数实验室的条件为:固定液5~10ml,照射10~30s,照射后固定液的温度≤50。C。其方法为:将实验标本置于微波炉旋转台的中央,周边放一烧杯盛300~500ml纯水,吸收照射时炉内产生的热量,选择强挡照射10~30s 。接通电源(on)至微波产生利用超声波代替微波照射,或二者并用,照射后,组织标本和固定液的温度升高较少,亦能获得短时间内良好的固定效果(Yasuda 1992)。
二、切片
光镜ICC染色,常用的有冰冻切片和石蜡切片两种,二者各有其优缺点,应根据抗原的性质、实验室条件,合理选择之。对未知抗原显示时,最好同时应用。冰冻切片为ICC研究所首选。
Shi(1991)等报告:微波炉照射的切片,能够激活部分核内抗原活性。其机制不清,可能与高温、高热的作用,使核内DNA双链解开,DNA、RNA解离,抗原暴露有关。其步骤为:将切片脱蜡水洗后,置染色瓶中,加入去离子水或缓冲液至瓶颈处,反复多次照射,每次1min,照射5~10次,每次照射后,补充瓶中蒸发掉的液体,照射温度不超过90~95℃为宜。此处理后,细胞核染色以苏木精为佳(详见第一章 )。
第二节 染色方法
一、本科标抗体法
酶标抗体技术是通过共价键将酶连结在抗体上,制成酶标抗体,再借酶对底物的特异催化作用,生成有色的不溶性产物或具有一定电子密度的颗粒,于光镜或电镜下进行细胞表面及细胞内各种抗原成分的定位。
(一)酶的种类及特点
从理论上讲,用细胞化学方法能显示的酶,均可用于标记抗体,进行ICC染色,但实际上在ICC中所能用的酶并不多。现将常用的几种酶列于表4-1,供选用时参考。
表4-1 免疫细胞化学常用的酶
| 名 称 | 分 子 量 | 内 源 性 | 商 品 |
| Horseeradish peroxidase(E.C.1,11,1,7) | 40~45KD | + | 有 |
| Alkaline phosphatase (E.C.3,1,3,1) | 80~120Kd | ++ | 有 |
| Acid phosphatase (E.C.3,1,3,2) | 100Kd | +++ | 有 |
| Glucose oxidase | 160~190kD | - | 有 |
Sternberger(1986)指出,用于标记的酶应具备以下几点①酶催化的底物必须是特异的,且容易被显示,所形成的产物易于光镜或电镜下观察;②所形成的终产物沉淀必须稳定,即终产物不能从酶活性部位向周围组织弥散,影响组织学定位;③较易获得的酶分子,最好有商品出售;④中性pH时,酶应稳定,酶标记抗体后,保存1~2年活性不应改变,且酶的催化活性(Turnover)越高越好;⑤酶标过程中,酶与抗体连结,不能影响二者的活性;⑥被检测组织中,不应存在与标记酶相同的内源性酶或类似物质。
其中,①②两点甚为重要,因为并非容易显示的酶均能形成不可溶性的复合物。一般认为,辣根过氧化物酶(Horseradish peroxidase , HRP)较佳,是最常用的一种酶。HRP广泛分布于植物界,以植物辣根(西洋山嵛菜)的叶内含量最丰富而得名。它是由无色的酶蛋白和深棕色的铁卟啉结合组成的一种糖蛋白,糖占16%~18(8条糖链分布在HRP分子表面),分子量40kD,最适pH5~5.5,最适温度40~45℃; pH4~11,50℃以下均较稳定,易溶于水和58%以下的饱和硫酸铵溶液。酶的活性中心含铁卟啉,称辅基,最大吸收光谱为403nm,而其余非活性的酶蛋白部分吸收光谱为275nm,HRP的纯度是用二者的光密度比值(Od 403/275)衡量,Reinheit Zahl (RZ)表示。一般认为,标记酶的RZ值为3.0左右,不应小于2.8,RZ值越小,酶的纯度越差,例如RZ值为0.6的酶,含非活性的酶蛋白量高达75%。对于纯度低、质量差的酶,需纯化后使用。
除HRP外,碱性磷酸酶(Alkalinephasphotase, ALP)和葡萄糖氧化酶(Glucose oxidase, GOD)也较常用。ALP分子量约为HRP的2~3倍,最适pH9.0~9.5左右,比较稳定,内源性ALP也较易消除,大部分均可被左旋咪唑(Levamisole,分子量240.8kD)抑制,但肠粘膜表面的内源性ALP活性不受影响。目前所用的ALP大多系由牛的肠粘膜提取制得,所以肠粘膜等呈强阳性反应。
ALP最初是由Bulman等用于标记抗体的。选用不同的底物,可形成不同颜色的终产物,例如以萘酚(As-Mx)和快蓝(Fast Blue, FB)为底物,生成蓝色沉淀。用快红(Fast Red, FR)代替FB,形成红色不溶沉淀。与HRP/4-氯-1-萘酚(CN)或DAB形成的沉淀形成鲜明对比,但FB、FR等沉淀物溶于有机溶剂,不能进行脱水、透明等处理。据报告,利用新品红(New fuch-sine )显色,形成的红色沉淀产物不溶于有机溶剂,不褪色,轻度核复染后,可制成半永久性保存标本。
GOD所催化的底物为葡萄糖,电子供体为对硝基四唑蓝(P-Nitroblue Tetrazolium),终产物为不溶性的蓝色沉淀,比较稳定。从理论上讲GOD较ALP、HRP为佳,因为哺乳动物组织内不存在内源性GOD,但其分子量较大,具有较多的氨基,在标记时易形成广泛的聚合,影响酶的活性,故GOD主要用于ICC双重染色和两种酶的放大技术。
(二)本科标抗体的制备(Boosma,DM, 1983)
酶标抗体与荧光色素标记抗体不同,它需借助桥-偶联剂的作用,将酶连结在抗体分子上。偶联剂是一种双功能试剂,具备3个基本特征:①偶联剂与抗体和酶之间的连结,必须是不可逆的,即借共价键连结;②偶联剂不应影响酶和抗体的活性;③不能因偶联剂的加入,使酶与组织成分了生非特异结合。
在HRP标记抗体中,常用的偶联剂有戊二醛、过碘酸钠及Maleimide等,现简介如下。
1.戊二 醛标记法戊二醛为制备各种酶标抗体最常用的偶联剂。市售戊二醛往往含有戊二酸、丙烯释及戊二醛自身聚合本等杂质,故需纯化后使用。戊二醛的纯度用含杂质的二醛的单体戊二醛的OD比值表示,它们的最大吸收光波长分别为235nm 和280nm,二者的OD比值(235/280)小于3时,制备酶标抗体的效果较好,大于3时,需经蒸馏或Sephadex G-10柱层析或活性碳吸附等处理,除去杂质后应用。其制备方法分一步法和两步法;基本原理相同,是使戊二醛的两个醛基之一与酶蛋白的赖氨酸结合,另一醛基与免疫球蛋白上的氨基结合,将酶连结于抗体上。
(1)一步法:将酶、抗体、戊二醛按一定比例混合,经透析除去标记物中剩余的戊二醛,制得酶标抗体。优点是简单省时,缺点是反应程度不易被控制,因为酶蛋白分子和抗体蛋白分子同戊二醛间的反应速率不同,抗体蛋白的氨基数远较HRP为多,与戊二醛反应快,因此在戊二醛的作用下,抗体蛋白易通过分子内和分子间的彼此交联,形成较大的聚合体,而与酶蛋白分子间的交联相应减少,影响酶的标记。据Nakane等推算,加入的HRP仅20%与抗体连结,标记率较低(约1%~5%)很难获得理想的酶标抗体。
(2)二步法:首先用过量的戊二醛与HRP反应(HRP:戊二醛为1:105),以保证酶分子仅与戊二醛的一个醛基结合,另一个醛基游离;然后用层析法除去多余的戊二醛,制成活性HRP(HRP-戊二醛复合物),再加入过量的抗体,使活化HRP上剩余的醛基与抗体蛋白分子上的氨基结合,制成酶标抗体。过量的抗体可以保证酶与抗体间均匀连结,避免酶本身聚合。根据所用的HRP与抗体(IgG)比例不同,酶标记率各异,平均为5%~25%。标记步骤如下:
①10~15mgHRP(RZ=3.0),溶解于0.2ml 1.25%戊二醛中(0.1mol/L磷酸缓冲液配制),18h室温。
②透析或 SephadexG-25柱层析(0.15mol/l NaCl平衡),去除过量的戊二醛,收集活化HRP。
③浓缩活化HRP至10mg/ml左右,加入抗体5mg(1.0ml 0.15mol/L NaCl 溶解)。
④碳酸盐缓冲液(pH9.5)调整pH至9.0~9.5,使抗体与活化HRP结合,4℃24h。
⑤加入0.1ml赖氨酸缓冲液,阻断未反应的醛基,4℃,2h。
⑥用半饱和硫酸铵沉淀5次,对PBS透析24h,4℃,换3次PBS,除去硫酸铵(10000rpm/min,30min)。
⑦或用凝胶色谱法(SephadexG-200/Sephacryl S-200) 等分离标记抗体。
注意:该方法要求HRP的RZ值在3.0左右,游离氨基较少,与戊二醛反应后,制成的酶标抗体大部分为单体;而RZ值小于2.8的HRP,含有较多的游离氨基,与戊二醛反应后,易形成多聚体,使方法的敏感性下降。
2.过碘酸盐氧化法严格地讲,过碘酸钠(Sudium periodate)不是一种真正的偶联剂,其本身并非作为桥连结在抗体和酶之间,而是借助于过碘酸钠的氧化作用,将酶连结在抗体上。该方法仅适于含糖较丰富的酶(如HRP)的标记。我们知道,HRP分子的糖本身与酶活性无关,利用过碘酸钠氧化这部分糖分子内的-CH基,使之生成-CHO基,再与抗体蛋白的游离氨基反应,生成Shiff’s碱。此Shiff’碱在pH降低时呈可逆性解离,所以经氢硼化钠(NaBH4)还原,形成稳定的酶标抗体复合物(图4-1)。为防止生成的-CHO基与酶蛋白氨基自身交联,预先可用二硝基氟苯(Dintro-fluorobenzene)处理HRP,阻断分子内的ε-、α-氨基。
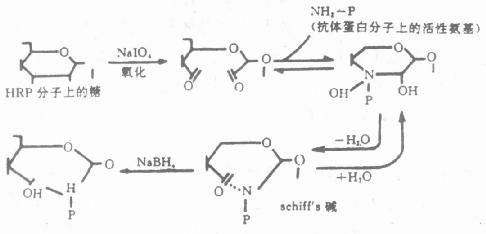
图4-1 过碘酸盐氧化原理
过碘酸盐氧化法,酶的RZ值≥3时较佳;RZ半乳糖
凝集素(Phytoagglutin, PNA),通常以其被提取的植物命名,如刀豆素A(Conconvalina,ConA)、麦胚素(Wheat germ agglutinin, WGA)、花生凝集素(Peanutagglutinin, PNA)和大豆凝集素(Soybean agglutinin, SBA)等,凝集素是它们的总称。凝集素不是来源或参与免疫反应的产物,它们之所以被收入本书,是由于凝集素具有的某些“亲合”特性,能被免疫细胞化学技术方法所应用。因此,Ponder(1983)提出应称“凝集素组织化学”而不能称为“凝集素免疫组织化学”。
一、凝集素的特性
凝集素具有多方面的特性,在此我们仅简要提及其与免疫细胞化学技术方法应用有关的某些特性。我们知道,生物膜中含有一定量的糖类,主要以糖蛋白和糖脂的形式存在。凝集素最大的特点在于它们能识别糖蛋白和糖肽中,特别是细胞膜中复杂的碳水化合物结构,即细胞膜表面的碳脂化合物决定簇。一种凝集素具有对某一种特异性糖基专一性结合的能力,如刀豆素与α—D—吡喃糖基甘露糖(α—D—Mannopyranosy)结合;麦芽素与N—乙酰糖胺(N—acetyl glucosamine)结合;菜豆凝集素与N—乙酰乳糖胺结合(见本章 表6—1)。 因此,凝集素可以作为一种探针来研究细胞膜上特定的糖基。另一方面,凝集素具有多价结合能力,能与荧光素、生物素、酶、胶体金和铁蛋白等示踪物结合,从而在光镜与/或电镜水平显示其结合部位。
二、凝集素的应用
一般认为细胞膜上特定的糖基可用以区别细胞的类型和反映细胞在分化、成熟和肿瘤细胞性变中的变化。仅在某些特殊的例子,其细胞结合凝集素的性能可以预先估计,如双花扁豆素之于血型A物质的特异性,荆豆凝集素之于血型O物质2—L—岩藻糖的特异性,然而在绝大多数情况下,关于由凝集素所识别的碳水化合物决定簇的种类,关于携带决定簇的分子的性质和机能,完全凭实验经验去发现。
1.作为细胞分化和成熟的标记应用凝集素作为细胞分化的标志,在这方面的应用报告最多,而且研究比较集中于血细胞,特别是淋巴细胞的分群。如Rose(1980)等发现在小鼠胸腺皮质内不成熟的T淋巴细胞呈PNA阳性反应,在小鼠小肠集合淋巴小结的生发中心也发现有20%左右的PNA阳性反应细胞,后者是否属于不成熟的T淋巴细胞,是值得进一步研究的问题。Newman等(1979)以荧光素标记凝集素PNA,发现在大鼠乳腺上皮的不同分化时期显示不同的荧光强度。在不成熟的大鼠乳腺上皮细胞,荧光弱或无,随着性成熟期到妊娠期乳腺上皮荧光程度逐渐加强,而泌乳期荧光强度达最高峰。在皮肤角质细胞自基底向表层分化、成熟的过程中,细胞表面的碳水化合物的分布和性质都在改变。Brabed等(1981)应用新生大鼠皮肤的实验表明,皮肤各层细胞分别与不同的凝集素相结合。麦芽素与角质化细胞相结合,蓖麻素与棘细胞和基底细胞相结合,而荆豆凝集素标记在棘细胞的表面。在成肌细胞(myeoblast)的分化与成熟过程中,Winaod 和Luzzati(1975)注意到类似的皮肤的改变。
2.作为细胞特殊类型的标记Kivela和Farkkanen(1987)发现在人视网膜,PNA标记视锥细胞而不标记视杆细胞。在乳腺、乳腺上皮细胞呈PNA阳性反应而肌上皮细胞和间质细胞呈PNA阴性反应。以多种凝集素对小鼠、大鼠和兔的肾组织切片进行染色结果表明,刀豆素A和蓖麻素存在于肾脏的各部,PNA和双花扁豆凝集素(DBA)主要分布于远曲小管和集合小管上皮细胞,荆豆凝集素(UEA)主要分布在血管内皮细胞,而麦芽素分布在肾小球。应用DBA对RIII和DDK品种的小鼠研究表明,DBA主要结合在各种组织内毛细血管内皮细胞上,电镜观察显示DBA结合在内皮细胞的表面,在趣的是在RIII品系小鼠某些组织的内皮细胞显示肯定的DBA阴性反应,说明同一种属动物的血管内皮细胞也存在有组织特异性的差别。Streit和Kreutzberg(1987)发现GriffoniaSimplicifolia凝集素特异性标记面神经节 内的小胶质细胞,其它类型的胶质细胞如星状胶质细胞(astrocyte)等都显示阴性反应。在切断面神经后,增殖的小胶质细胞对Griffonia Simplicifolia凝集素的反应加强,免疫电镜观察表明,凝集素主要沉积在细胞膜或小胶质细胞突起的轴膜表面,特异性结合糖基是α—D—半乳糖。上海医科大学附属肿瘤医院免疫病理室应用12种凝集素(表6-1)对人胚胎及各种正常组织进行了系统的凝集素受体的定位研究,结果表明,凝集素受体的分布并无即定规律可寻。如胃粘膜主细胞为PNA受体,而壁细胞为BSL受体,双花扁豆受体(DBA)主要出现在大肠部份。
3.在肿瘤中凝集素结合的改变肿瘤细胞伴有细胞膜的改变,细胞膜上的糖基也会产生相应的变化,可用凝集素检测出来。大量研究发现,凝集素可作为肿瘤组织源性的标记、肿瘤特异性诊断的标志、肿瘤恶性的标记和不同肿瘤的分化标记。如张华忠等(1987)报道115例胃癌标记PHA阳性率高达90.43%,而正常胃粘膜基本是阴性,故认为PHA是胃癌的诊断性标志。BSA对乳腺恶性肿瘤阳性率达79%,而对良性病变均呈阴性反应,提示BSA可能为乳腺恶性肿瘤的相关标志。凝集素还有助于判别肿瘤的组织类型,如神经系统星形细胞瘤ConA阳性,小胶质细胞瘤阴性,肾腺癌UEA1阴性,透明细胞癌阳性。
三、凝集素在免疫细胞化学中的应用
凝集素可为荧光素、酶和生物素等所标记,分别进行下列染色法:
1.直接法标记物直接标记在凝集素上,使之直接与切片中的相应糖蛋白或糖脂相结合。
(1)切片脱蜡至水。
(2)凝集素标记物(100μg/ml),室温,30min。
(3)TBS洗3次,每次2min。
(4)如为荧光素标记物,封片用荧光显微镜观察。如为酶标记物,则应依次进行呈色、脱水、透明和封固后在光学显微下观察。
直接法的优点是简便,目前商品用的凝集素药盒已能购得。但灵敏性不够高。
2.间接法将凝集素直接与切片中的相应糖基结合,而将标记物结合在抗凝集素抗体上。
(1)脱蜡至水。
(2)用含3%的H2O2的甲醇阻断内源性过氧化物酶10min。
(3)凝集素稀释液(100μg/ml)孵育30min。
(4)TBS洗3次,每次2min。
(5)用标记了的抗凝集素抗体(1:100)孵育30min。
(6)TBS洗3次,每次2min。
(7)呈色、脱水、透明、封片。
(8)观察。
间接法染色还可进一步改良为:①三步法:即在凝集素孵育后,接着用抗凝集素抗体孵育,再用标记了的抗-抗凝集素抗体孵育,层层放大,进一步提高其敏感性,PAP复合物也可作为标记物标记在抗-抗凝集素抗体上。②抗生物素—生物素凝集素法:用结合了生物素的凝集素孵育切片后,TBS洗后再以抗生物素—标记物与之结合。间接法较直接法和直接法敏感性高5~10倍或更多一些,但必须购买或自制抗凝集素抗体。
3.糖—凝集素—糖法本法是利用过量的凝集素与组织切片中特定的糖基相结合。经冲洗后,凝集素上还存在未被占用的结合部位,将这些部位与有过氧化物酶标记的特异性糖基相结合,形成一个三明治样的糖—凝集素—糖的结合物。
(1)脱蜡至水。
(2)用含3%的H2O2的甲醇阻断内源性过氧化物酶10min。
(3)用100μg/ml的凝集素孵育30min。
(4)TBS洗3次,每次3min。
(5)用100μg/mlHRP标记的糖液孵育30min。
(6)TBS洗3次,每次3min。
(7)DAB呈色、脱水、透明、封固。
本法特异性强,灵敏度高,因为这不象生物素—抗生物素法那样要改变凝集素,又不需要像抗体那样要制备抗体。HRP本身含有甘露糖,能与刀豆凝集素A、扁豆凝集素和豌豆凝集素结合。但对其它的凝集素,本法目前普及还有一定困难,因为要将过氧化物酶结合到其它凝集素上,就需要将一个适当的碳水化合物基团嵌入过氧化物酶,称为糖基化(glycosylation),方法虽不复杂(Lee 等1976),但需一定的试剂和设备。商品化能提供的糖基化过氧化物酶品种尚有限。
【注意事项】
①凝集素需要重金属离子维持其活跃的结合部位,如果金属离子耗尽了,就会影响凝集素的结合能力。因此,有作者主张用TBS作为缓冲液,内加微量的金属,配方是:Tris 60.57g,NaCl87g,H2O加至1000ml,其中含CaCl2、MgCl2各1.0mmol/L。或在进入凝集素孵育前,先用该液孵育,以增强凝集素结合力。
②和其它抗体血清应用一样, 应用每批新的凝集素实验时,都先要用缓冲液稀释成不同等级;如8,16,32,64,125,250,500,1000μg/ml,经染色选择最佳稀释度。
③有作者认为阻断组织内源性过氧化物酶所用的H2O2对碳水化合物有影响,可能改变凝集素的结合形式,因此,应尽量少用或不用,我们及一些作者在实验过程中发现影响不明显。
④有作者认为凡经固定的组织切片,不论是石蜡包埋切片或冰冻切片,都有可能使组织中抗原隐蔽,为了暴露隐蔽了的碳水化合物基团,主张在凝集素孵育前用酶处理切片,常用胰蛋白酶液(配制见附表)进行适当的孵育。
⑤已知哺乳动物的质膜含有占蛋白总量的1%~10%的碳水化合物,它们以低聚糖(oligosacchride)糖脂,并主要以糖蛋白的形式存在,糖蛋白线性的或分支的旁链可能含有两个到多个单糖残基,通常有两种或更多的单糖,在单糖单位末端常常是一个带负电荷的N-乙酰神经氨酸的残基,一个唾液酸(siallic acid)。有作者在凝集素实验中常用神经氨酸酶(neuramidinase)分解细胞表面的唾液酸或神经氨酸,以暴露出隐蔽的能与聚集素结合的次终末的碳水化合物。配制方法是将神经氨酸酶(Type V,Sigma Lot 63F—8172 63F--8172)用醋酸缓冲液(含2%牛血清白蛋白)BSA配成0.5μg/ml。
切片脱蜡后进行酶消化的过程中,将该组织切片置于上述配制液中,在湿盒内,37℃孵育30min。用缓冲液洗后,进行凝集素染色。
⑥对照试验,和其它组化染色一样,凝集素染色也需要设对照试验(最好在相邻切片进行)。由于凝集素具有单糖特异性,如果外加相应的糖,把凝集素的结合部位占有了,凝集素就不能再与组织中的糖基相结合了。一般采用的方法是将凝集素预先与相应的糖(0.2mol/L)在室温孵育30min,使之占有凝集素结合部位,再将此液代替凝集素进行孵育,结果应为阴性。在某些情况下即使提高糖液的浓度也不能达到完全的抑制。这时,只有用与凝集素有高亲合力的低聚糖(oligosachrides)代替或将切片预先用相应的糖苷酶孵育去除特异性糖基(Watanalte等,1981)。
四、HRP标记凝集素及凝集素抗体的制备
1.HRP标记凝集素法适用于小量的凝集素标记(Ponder 1983)。
(1)HRP的活化
①将10mg HRP(SigmaType VI)溶解在1ml 0.3mol/L 的重碳酸钠溶液中(1.25g/50ml)。
②加50μl含1%氟二硝基苯的无水乙醇溶液,室温轻度搅拌1h。
③加1ml含0.06mol/L的过碘酸钠的蒸馏水(0.62g/50ml),室温下轻度搅拌30min。
④在室温下加两滴0.16mol/L乙二醇,轻搅1h。
⑤用Sephadex G—25装成小柱,用0.3mol/L的重碳酸钠溶液,使含HRP的混合液过柱。
(2)结合
①正常条件下,可用凝集素及过氧化物酶各半量,但其比例可根据需要调整,以10mg凝集素溶解在25ml的重碳酸盐缓冲液里,加到“活化”的HRP中去 ,这样得出的凝集素浓度大约是0.3mg/ml,可加进0.1mol/L相应的特异的糖,以保护凝集素结合时的结合部位。糖在下步透析或层析时去掉。
②在室温下轻搅拌,结合3h。
③加入1mg硼氢化钠,室温下作用1h以稳定结合物活性。
④用1N HCl调整pH至6.4,留置室温下过夜。
(3)纯化
①用适当的凝胶层析柱如SephadexG—200、Saphacryl—300层析法将游离的凝集素、游离的过氧化物酶和高分子量的成分从结合物分离出去,包括抑制性的糖也在此时分离出去。
②提纯后的结合物在280nm和403nm波长处测量其吸光率。用1%BSA先通过0.22μm的微孔过滤器,以确定被结合的凝集素是否被吸附在滤器中,然后上述结合物经过该微孔滤过器过滤到一个无菌瓶中。
③加入叠氮钠,浓度1:1000,保存于4℃C备用。
2.抗凝集素抗体的制备抗凝集素抗体的制备法与一般免疫血清制备大致相同(见第二章 )。所不同的是:①凝集素具有较强的毒性,易使被免疫动物发病或甚至死亡。解决的方法是经处理使凝集素变性,从而减低其毒性,但同时要保证其抗原性不受或少受破坏。②要设法阻断凝集素的糖结合部位,以减少对这些部位的抗体的产生,解决的办法是预先以相应的糖阻断这些结合部位。Leathem 和Atkin(1982)设计了一个办法,他们首先应用琼脂糖(Sepharose)珠,珠上有一系列共价糖的附着,这些糖可阻断凝集素结合部位,这种糖珠有商品供应(Sigma LtD),每1ml糖珠能结合大约12mg凝集素。其次用加入福尔马林和加热的方法使凝集素变性,毒性减低而不影响抗原性。具体操作如下:
①1ml琼脂糖—半乳糖珠与1mg花生凝集素相结合,室温1h。
②加入10ml 10%福尔马林,在水浴中加热到60℃,1h。
③用盐水离心洗珠,将珠分成以10μl为单位的若干小份,保存在-20℃。
④皮下多处注射两只家兔背部,每次注射100μl,每隔三周注射一次。取血40~50ml,保存在-20℃备用。
第四节 免疫细胞化学技术的某些新进展
免疫细胞化学技术在继续改进和完善中,新的技术方法不断出现。除目前国内已开始应用的免疫金技术和免疫金银技术外,80年代,新的免疫细胞化学技术还有半抗原交联抗体法和令人瞩目的分子杂交免疫细胞化学技术。免疫金银技术和分子杂交免疫细胞化学技术将分别以专章 叙述,本节 仅就半抗原交联抗体法作一简要介绍。
【半抗原交联抗体法(Hapten –Coupled techniques)】
近年来,为提高敏感性和减低非特异性染色,有些学者(Cammisuli 1976; Jassani 1981;Falini 1983)在常规免疫细胞化学技术的基础上,发展了半抗原交联抗体法,可作免疫荧光或免疫酶染色。本法的基本原理是应用半抗原标记第一抗体。常用的半抗原有阿散酸,即对氨基苯砷酸(Arsanilic acid, ARS),对氨基苯酰甘氨酸(P—aminobenzoyl glycine),亚对氨苯酰甘氨酸(N—P—aminobonozyl glutamic acid)和二硝基苯氨基丙睛亚胺酸脂(dinitrophenyl aminopropionitrileimido ester, DNP)。通过酰胺化反应把半抗原结合到抗体分子上。在间接法(二步法)中,先以半抗原标记的特异性抗体(第一抗体)孵育切片,然后加入半抗原的标记抗体,(标记物可为荧光素或酶)(图6-4)。在PAP染色(三步法)中,先用半抗原标记的第一抗体孵育切片,再加未标记的抗半抗原特异性抗体作为桥抗体,第三步加半抗原交联的PAP与之反应,并作酶的呈色反应(图6-5)。第一和第三抗体分别为半抗原标记的特异性抗体和以半抗原标记的抗酶抗体制备的含半抗原的PAP复合物。本法具有许多优点,主要是:①敏感性高。由于本法是通过酰胺化反应标记半抗原,对抗体活性损失极小,抗体保留较高的活性,能结合较大量的半抗原,平均每个球蛋白分子可同时结合20个半抗原分子,每个半抗原又可用抗半抗原抗体相结合,特异免疫反应明显放大,其敏感性明显高于常规的免疫酶和免疫荧光染色技术,特别有助于发现滴度低的同种抗血清和含抗原量较少的组织。②可用于双重免疫染色。利用两种交叉反应的半抗原如阿散酸和对氨基苯酰甘氨酸分别标记来自同一种动物的两种特异性(第一抗体)血清,应用两步法或三步法,即可在同一张切片内同时显示两种不同的抗原。③由于特异性强、敏感性高,因此背景染色低。
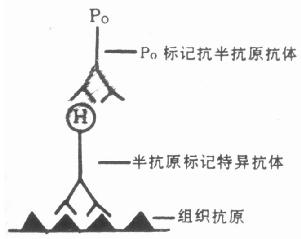
图6-4 半抗原标记抗体酶技术—两步法
图6-5 半抗原标记抗体酶技术—PAP法
参考文献
1.Bayer EA, et al .Affinity Cytochemistry:The localization of lectin andantibody receptors on erythrocytes iva avidin—biotin complex.FEBS Letters, 1975;68:240
2.Brabec RK, et al .Differential lectin binding tocellular membranes in the epithelium of the new born rat.Proc Natl Acad Sci USA, 1987;77:477
3.Falini B. New development inimmunoperoxidase techniques and their application.Arch Pathl Lab Med, 1983;107:105
4.Jasani B. Et al. Use of monoclonal antihaptenantibodies of immno—Iocalization of tissue antigens.J Clin Pathol, 1981;34:100
5.Kivela T and Tarkanen A.A lectin cytochemical study ofglycoconjugates in the human retina.Cell Tissue Res, 1987;249:277
6.Newman RA, et al .Binding of peanut lectin to breastepithelium, human carcinoma and cultured rat mammary stem cells:Use of the lectin as a marker ofmammary differentiation.J Natl Cancer Inst, 1979;63:1339
7.Ponder BAJ, et al .Lectin histochemistry. (Immunocyto—chemistry Eds.JM Polak and SV Noorden )WrightPSG Bristol London Boston 1983;129
8.Rose ML, et al .Peanut lectin binding propertiesof germinal centres of mouse lymphoid tissue.Nature, 1980;284:364
9.Streit WJ and Kreutzberg GW.Lectin binding by resting andreactive microglia. Neurocytology, 1987;16:249
10.Virtanen AL, et al .Fluorochrome – coupled lectinsreveal distinct ceullar domains in human epidermis.J Histochem Cytochem, 1986;34(3):307
11.Watanalte M, et al.Discrete distribution of bindingsites for Dolichos biflrous agglutinin and for peanut agglutinin (PNA)in mouseorgan tissues.J Histochem Cytochem, 1981;29:779
12. 王益寿.葡萄球菌蛋白A的性质和应用.国外医学(生物制品分册),1980;4:145
13. 张华忠,等. 凝集素在人体正常组织的定位.中华医学杂志,1985;65:144~147
14. 沈铭昌,等.应用DBA标记胃粘膜上皮化生与胃癌的关系.肿瘤,1987;7:55~57
15.Ueda T, et al.Lectin histochemistry of maligantfibrohistocyte tumors.Am J Surg Pathol, 1987;11(4):257
16.Ree HJ.Lectin disfinction of benign frommalignant histocyts.Cancer, 1985;56(8):2046
第七章 电子显微镜免疫细胞化学技术
第一节 电子显微镜免疫细胞化学技术概述
免疫细胞化学技术为在细胞水平上研究免疫反应做出了贡献,但由于光学分辨率的限制,不可能从细胞超威结构水平观察和研究免疫反应。因此,Singer于1959年首先提出用电子密度较高的物质铁蛋白(ferritin)标记抗体的方法,为在细胞超威结构水平研究抗原抗体反应提供了可能。在此基础上,相继发展了杂交抗体技术、铁蛋白抗铁蛋白复合物技术、蛋白A-铁蛋白标记技术、免疫酶技术及胶体金技术等。电子显微镜免疫细胞化学技术(以下简称免疫电镜技术技术)区别于免疫细胞化学和常规电镜技术主要在以下几方面:
一、组织固定与取材
在这方面的要求是即要保存良好的细胞超威结构,又要注意保持组织的抗原性。因此,选用固定剂不宜过强。常用的免疫电镜固定剂有多聚甲醛—戊二醛混合液和过碘酸-赖氨酸—多聚甲醛液(Periodate—Lysine –Paraformaldehyde简称PLP液)。也有采用Bouin氏液、Zamboni氏液或4%多聚甲醛液(其配制法见附录)。国外不少文献推荐应用PLP液于免疫电镜技术,认为该固定液对含糖类丰富的组织固定效果特佳,因为组织抗原绝大多数由蛋白质和糖两部分组成,抗原决定簇位于蛋白部分,有选择性地使糖类固定,就可使抗原性稳定。PLP液中过碘酸能氧化糖类,使其产生醛基,再经赖氨酸作用,使新形成的醛基分子间和分子内相互连接,稳定组织抗原。但赖氨酸价格较贵,不如多聚甲醛戊二醛固定液经济、简便、效果佳。在取材方面,免疫电镜技术较光镜免疫化学技术要求更迅速、精细。
二、免疫染色
分为包埋前染色、包埋后染色和超薄切片免疫染色三种。
1.包埋前染色即先行免疫染色,在解剖显微镜下将免疫反应阳性部位取出,修整成小块,按常规电镜方法处理,经锇酸固定、脱水、包埋。如果特异性免疫反应的范围太小,为了准确定位,可作第二次包埋,即第一次包埋时将组织置于两层thermanox塑料片之间,中夹环氧树脂如夹心面包式,进行高温聚合,然后在解剖显微镜下取出需要部位作第二次包埋。包埋前染色的组织,以中层较为理想。表层因受机械修整,结构往往保存不好,深层因抗体不能透入,免疫反应弱或无。在作超薄切片前应先切半薄切片,寻出免疫反应阳性部位。根据作者经验,半薄切片可在相差显微镜下不染色进行观察(指PAP染色法),免疫反应部位呈黑点状。在HE或甲苯胺蓝染色的半薄切片上,免疫反应部位呈棕黄色。据此定位作超薄切片,可大大提高阳性反应检出率。为避免电镜铅、铀染色反应与免疫反应之间的混淆,可取相连续的起薄切片,分别以两个铜网捞取,其中之一进行染色观察,另一以铀单染色或不染色进行对照观察。
包埋前染色法的优点是:①切片染色前不经过锇酸后固定、脱水及树脂包埋等过程,抗原未被破坏,易于获得良好的免疫反应。②可在免疫反应阳性部位定位作超薄切片,提高电镜下的检出率。特别适用于含抗原量较少的组织,但由于经过一系列的免疫染色步骤,常出现一定的超微结构损伤。
2.包埋后染色组织标本经过固定、脱水及树脂包埋、制成超薄切片后,再进行免疫组化染色。由于是以贴在网上的超薄切片进行免疫染色,故又名之载网染色(on grid staining)。必须指出的是:①后固定中是否应用四氧化锇存在不同意见,作者经验一般以不用四氧化锇为佳,或尽量缩短在四氧化锇中停留的时间。有作者认为,从理论上讲,四氧化锇具有保存抗原的作用,但实践证明应用四氧化锇可使抗原活性明显减低。②在载网染色过程中,铜网易与化学物质产生反应,故需选用镍网或金网。③在免疫组化处理的全过程中,应注意保持网面的湿润,网面干燥会影响抗体活性。本法的优点是超微结构保存较好,方法简便,阳性结构有高度的可重复性,还能在同一张切片上进行多重免疫染色。但抗原活性在电镜生物样品处理过程中可能减弱甚至丧失;环氧树脂中的环氧基,在聚合过程中可能与组织成份发生反应而改变抗原性质;包埋在环氧树脂中的环氧基,在聚合过程中可能与组织成份发生反应而改变抗原性质;包埋在环氧树脂中的组织不易进行免疫反应等。因此,免疫组化工作者曾试图以不同的方法如饱和苯溶液,无水酒精中NaOH饱和溶液或乙氧化钠溶液等以减少或去除包埋剂,取得不同程度的效果。现普遍采用的是在进行免疫染色前,以H2O2液蚀刻数分钟,以去锇和增强树脂的穿透性。
3.超薄冰冻切片按照Tokuyasu建立的方法,将组织置于2.3mol/L蔗糖液中,以液氮速冻,在冰冻超薄切片机上切片,切片厚度可略厚于常规树脂切片。冰冻超薄切片由于不需经固定、脱水、包埋等步骤,直接进行免疫染色,所以抗原性保存较好,兼有包埋前和包埋后染色的优点。
三、包埋
(一)树脂包埋
国内现普遍采用的是环氧树脂包埋法,可直接脱水后包埋,也可将小片组织或半薄切片贴在载片上,将充满环氧树脂的明胶囊倒置于切片上聚合、硬化,进行原位包埋。
(二)低温包埋
常规树脂包埋由于需高温聚合等处理程序,组织抗原性可能全部或部分丢失。因此,在免疫电镜技术方面,国外不少实验室已开始采用低温技术如低温包埋和冰冻超薄切片等,后者需配备冰冻超薄切片机,且技术难度较大,不如低温包埋法易推广。低温包埋剂的研究开始于60年代,80年代免疫细胞化学技术在电镜水平上的广泛的应用,为低温包埋剂的实验研究开辟了广阔的领域。国内已有较多的应用报道,国内一些实验室已开始摸索。作为低温包埋剂的多为乙烯系化合物如乙二醇甲基丙烯酸酯(Glycolmethacrylate,GMA),Lowicryls, LR White和Lr Gold等,目前国外生产厂家有Polysciences INC , Reichert—Jung 和LKB等系列产品。现将常用的几种低温包埋剂及其应用简单介绍如下:
1.Lowicryls 是丙烯酸盐(acrylate) 和甲基丙烯酸盐(methacrylate)化学物质,包括K4M、HM20、K11M、KM23等系列产品(Polysciences INC),其特点是能在低温下保持低粘度(K4M:--35℃;HM20:-70℃;K11M、HM23:-60℃~-80℃)和具有在光照射(紫外光,波长360nm)下聚合的能力,它的光聚合作用与温度无度。其中K4M和K11M具有亲水性,特别适合于免疫细胞化学的应用,因它能较好地保持组织结构和抗原性,减少背景染色。HM20和HM23具疏水性,能产生高反差图像,适用于扫描、透射电镜和暗视野观察切片的制作。所有这些种类的低温包埋剂都适用于冰冻置换技术。K4M的应用和报道较多,现侧重介绍如下:
(1)包埋剂的配制:商品提供的Lowicrys包埋剂由三个部分组成:单体(Monomer),交联剂(Crosslinker)和引发剂(Initator)。调整单体和交联剂的比例,增加交联剂的量,组织块的硬度增加。中等硬度的组织块,其配制比例如下:
K4M:单体 17.30g
交联剂2.70g
引发剂0.10g
K11M:单体19.00g
交联剂1.00g
引发剂0.10g
作者的经验,可用微量注射器加针头抽取后,注入棕色的玻璃容器避光,用玻棒轻搅3~5min或用一小管通入液氮气泡以搅拌之。勿过分搅拌,以防氧的气泡进入包埋剂中。
(2)生物样品处理程序(Lemanski 等1985)
①动物麻醉取材,以多聚甲醛—赖氨酸—过碘酸钠在9℃固定2h。
②磷酸缓冲液含7%蔗糖,pH7.2,冲洗过夜,0℃
③0.1mol/L 磷酸缓冲液,pH7.2,冲洗,0℃
④脱水:65%乙醇,1h ,0℃
80%乙醇,2h,-35℃
LowicrylK4M:80%乙醇=1:11h -35℃
LowicrylK4M:80%乙醇=2:11h -35℃
100%Lowicryl K4M 1h -35℃
100%Lowicryl K4M 过夜-35℃
⑤包埋:新鲜K4M置于胶囊内,将组织移入,在-30℃~-40℃以紫外线灯波长360nm 2× 15W(Ladd Research Industries Burlington VT)相距30~40cm照射24h使之聚合。如为100W灯泡,照射距离应大于85cm。聚合后的胶囊移至室温在紫外线下继续照射2~3天,可增加其硬度,便于切片。
(3)免疫染色
①切片(厚50~70nm)贴在金网或覆有碳膜的镍网上。所有下列步骤在室温、湿盒内进行。所有溶液需经微孔滤纸(0.25~0.45μm)滤过。
②正常羊血清30min。
③第一抗血清(PBS稀释),37℃2h。
④可用烧杯法(或塑料壶喷洗法),以镊夹镍网在第一烧杯中洗荡30min,然后在第二烧杯中洗荡1h 。
⑤正常羊血清30min。
⑥第二抗血清(胶金标记抗体)以正常羊血清稀释为1:1,以镍网置于血清滴上孵育1h。
⑦冲洗如④。
⑧覆于2%OSO4水溶液上,30min。
⑨冲洗如④。
⑩干燥后在电镜下观察。
(4)Lowicryl K4M快速包埋染色法(Altman等1984)
①包埋剂的配制:单 体13g
交联剂2g
引发剂75mg
②生物样品处理:除了聚合这一步骤外,下列所有步骤都在20℃进行。
1)组织用3%戊二醛—3%多聚甲醛磷酸缓冲液,pH7.4,在20℃固定1~2h。用磷酸缓冲液清洗后进行脱水。
2)脱水:在50%、75%和90%的双甲基甲酰胺(Dimethylformamde,DMF)内系列脱水,每步10min。
3)浸透:Lowicryl K4M:DMF=1:2 10min
LowicrylK4M:DMF=1:1 15min
100%Lowicryl K4M 20min
100%Lowicryl K4M 25min
4)包埋与聚合:组织移入装满K4M的胶囊中,以紫外线灯照射聚合(紫外线灯条件同上),灯和组织距离10cm,4℃照射45min,组织块在室温进行超薄切片(切片时水槽内水面应略低以防浸湿组织块的切面)。
5)以覆有碳膜的镍网捞取切片。
6)免疫染色。
整个包埋时间仅需4~5h。
Lowicryls应保存在暗处,-4℃,该物质有刺激性,在配制时应戴手套,以免触及皮肤。在通风橱内操作,以免蒸气刺激眼睛。如触及皮肤和眼睛,应立即以水冲洗局部,氧化重金属如KmnO4能与包埋剂作用而影响染色效果,以不用为佳。
2.LR White 和Lr gold 是一种混合的丙烯酸单体的透明树脂,具有极低的粘度(8cps)和较强的嗜水性,因此有较强的穿透性,有利于抗体(或抗原)和免疫化学物质穿过LR树脂,达到组织结合部位。在免疫细胞化学的光镜(半薄切片)和电镜水平应用都具有良好效果。标本脱水至70%乙醇即可,能较好地保持抗原性。Lr white和Lr gold在国外提供厂家有 Poly-sciences INC等,Lr gold是一种光引发低发低温聚合的包埋剂,对于免疫细胞化学特别适用,能最大限度地保持组织的抗原与抗体活性,其最佳光聚合温度在-25℃,在聚合后呈现金黄色,因而得名。Lr white可在热和冷两种情况下聚合,热聚合在60℃,24h,冷聚合在-25℃,需加加速剂(accelerator)调整配制比例。生物样品处理与免疫染色等同常规树脂切片。
3.GMA 是乙二醇甲基丙烯酸酯(Glycol Methacrylate 即2—hydroxyethyl methacrylate, HEMA)的缩写。远在60年代,电镜工作者就试图将其作为生物包埋剂应用于光镜和电镜。GMA作为电镜包埋剂的优点是电子密度大,影像反差好。但存在三个主要缺点:一是包埋聚合后的组织块很脆,不易修整和切片。二是聚合过程中易造成组织损伤如人为的细胞器肿胀。三是缺乏稳定性,不能承受电子束的轰击,包埋剂遇热升华,造成组织塌陷变形。故后来为环氧树脂所取代。聚合后的环氧树脂有良好的塑料稳定性,能承受电子束的轰击不变形,而且影像反差好,分辨率高,但其半薄切片染色不够满意始终是个有待解决的问题。而GMA包埋切片的染色效果明显优于环氧树脂。于是电镜工作者如Leduc和Bernhard(1986)尝试以增加一定比例的增塑剂如甲基丙烯酸酯和少量水外,并加入适量的增塑剂如聚乙二醇400(PEG400)以改变其硬度,加入适量的闻联剂乙烯二甲基丙烯酸酯(ethylene dimethacrylate)以增强其抗电子束轰击的稳定性。为避免聚合时过快,产生高温,损伤组织结构,选用低温型引发剂—过氧化苯甲酰(Benzoyl Peroxide),温度范围-10℃~-30℃。经过不断的配制改进,现GMA已广泛应用于半薄切片(1~3μm)的光镜观察,特别是组织化学方面的研究和电镜水平的免疫细胞化学技术。现将GMA包埋剂的配制、生物样品处理和电镜水平的免疫细胞化学染色方法简介如下:
(1)GMA单体溶液在出厂时都加有氢醌类稳定剂,用前须以每25ml单体溶液加一匙活性碳,在振荡器上振荡5min,过滤以除去氢酯,以免影响聚合。
(2)包埋剂的配制:
a. 100%GMA66.5ml
N—甲基丙烯酸丁酯 28.5ml
5%乙烯二甲丙烯酸酯 5.0ml
1.5 %过氧体苯甲酰 1.0g
1.0%PEG4001.0ml
b.A液:
GMA单体液 90ml
PEG 400 5~9.4ml
过氧化苯甲酰0.2~0.69g
搅拌溶解后置棕色瓶内; 4℃保存。
B液:
PEG 400 20ml
二甲基苯胺1ml
应用时A:B按10:1比例充分混合(张承志等1986)。
(3)生物样品处理:组织固定可用PLP液或1%戊二醛溶液(磷酸缓冲液配制,pH7.4)。经系列酒精脱水至新鲜的GMA单体溶液中浸24h。以上步骤可在室温或4℃进行。
(4)包埋聚合:组织移入盛潢包埋液的胶囊内,先放在真空泵内以去除包埋液中的气泡,然后在4℃,以紫外线灯(波长360nm)在距胶囊底部10~20cm处进行照射12~16h,以手指捏胶囊试其硬度可知聚合是否完成。在聚合完成后,将胶囊丢入热水中以去除胶囊外壳。
(5)切片贴在镍网上,按PAg技术进行包埋染色,其区别于EPON包埋剂者,在于GMA包埋切片染色所需时间较短,在第一抗血清中,GMA室温只需孵育1h,而EPON包埋切片需16~20h;在第二抗血清,即Pag 复合物中室温30min,而EPON包埋切片需1h。铀、铅双染后,电镜观察。
低温包埋剂常用于铁蛋白或胶体金免疫电镜技术的包埋后染色。能检出应用环氧树脂包埋难以检出的多种抗原。
四、对照试验
为确定方法的特异性,免疫电镜技术也需进行对照试验(同第一章 )。
总之,不论哪一种免疫电镜技术都面临微细结构的保存和组织中抗原活性的保存这一对矛盾,如戊二醛、锇酸等固定液有利于微细结构的保存,但对抗原活性有影响,而H2O2能增加树脂穿透性,但对微细结构有损伤,能使反应部位产生孔洞。在生物样品处理过程中,应同时注意到这两个方面。其次,每次免疫染色中的清洗工作应注意彻底,否则非特异性产物和其他污染物会影响特异性反应产物的显示和观察。根据作者经验,以塑料水壶加锥形喷水头喷洗,与镍网面成平行方向,即顺网面喷洗,较之目前通用的杯水洗涤法易于达到清洁目的。冲洗的残留水滴以滤纸吸干时,应注意不要触及载网本身。可将滤纸剪成三角形,以尖端接触水滴,即可达到吸干水份的目的,整个过程中,必须应用双蒸水,容器应专用。
第二节 免疫铁蛋白技术
一、基本原理
铁蛋白是一种含铁约占23%的蛋白质,分子量460kD,直径10~12μm。抗体与铁蛋白通过低分子量的双功能试剂结合为一种双分子复合物。此复合物既保留抗体的免疫活性,同时因为铁蛋白含有致密的铁离子核心,铁胶粒直径核心为55~60nm含2000~3000个铁原子,分布于四个区域,形成四个圆形致密区,具有很高的电子密度,便于电镜观察。铁蛋白来自许多动物,以肝、脾含量较高,其中马脾脏含量最高。因而,商品铁蛋白主要是从马脾脏中提取的。
二、铁蛋白的提取和纯化
取健康的马脾(新鲜或冰冻均可),去除脾外的淋巴结和脂肪组织,按湿重1:1.5或1:2加入蒸馏水,用组织匀浆器将组织匀浆,置水浴中加热至75~85℃,使铁蛋白以外的蛋白质变性,冷却后用2~4层纱布过滤,滤液离心取上清液,每100ml上清液中加入35g硫酸铵,充分搅拌使铁蛋白沉淀析出,4℃过夜,3000~10000r/min离心20min,弃去上清液,刮取沉淀物置透析袋内,剩余沉淀物以蒸馏水洗后,一并加入透析袋内对水透析,除去硫酸铵。100ml中加入4~5g硫酸镉使炎结晶,在室温或4℃冰箱内使之出现铁蛋白结晶。此铁蛋白结晶以2%硫酸铵(pH5.58)溶解后,如上述再用硫酸镉使之结晶。反复溶解,结晶六次,达到纯化。纯化后铁蛋白在半饱和的硫酸溶液内可保存1~2年。用时以蒸馏水透析除盐后即可应用,应用前可以用负染色法在电镜下观察,了解铁蛋白的完整性和纯度。
商品制备的铁蛋白是用2%硫酸铵溶液稀释的1%~2%溶液(pH5.85)。为了在应用中得到满意的结果,应用前必须将商品制备的铁蛋白进一步纯化。纯化的方法是用0.1n NaOH或0.1n HCl调整pH值至5.85,然后加入20%硫酸镉溶液使其在铁蛋白液中最终浓度为5%硫酸镉,此溶液置于4℃冰箱内2h(或过夜)至结晶完成,离心1h(2000r/min)充上清液。铁蛋白结晶再溶解,离心除去不溶性颗粒,倾出上清液,加入5%硫酸镉再结晶,至在显微镜下呈棕红色铁蛋白结晶为止。将结晶溶于2%硫酸铵溶液中,用50%5硫酸铵溶液沉淀三次,第三次沉淀形成的沉淀物溶于少量蒸馏水中,先对自来水透析,再对0.05mol/L,pH7.5磷酸缓冲透析12~24h。纯化的铁蛋白溶液用100000r/min 超速离心2h ,除去3/4无色上清液,沉淀物(内含铁蛋白)在4℃过夜,待完全融解后,用微孔滤过器(Millipore filter, 孔径 0.25~0.45μm)过滤,保存1~2年内仍可使用。
三、铁蛋白与免疫球蛋白的结合
一般用低分子量的双功能试剂把两者联结起来,常用的双功能试剂有间苯二甲基二异氰酸盐(简写XC)。甲苯2,4二异氰酸盐(简写TC)。邻茴香胺(简写BDD);对,二氟一间,间,二硝基二苯矾(简写FNPS)和戊二醛。近年来,普遍认为戊二醛作为联结剂效果较好,对抗体活性影响小,标记抗体产量高。分为一步法和二步法,现简介如下:
1.一步法以15mg 铁蛋白和3mg球蛋白溶于0.9ml 0.1mol/L磷酸缓冲液中,pH7.0,加入0.1ml新鲜配制的戊二醛溶液,使其最终浓度为 0.005%~0.05%, 加入0.02%叠氮钠(NaN3)防腐,此混合液置37℃24h,无需搅拌,结合完毕后加0.01mol/L赖氨酸中止反应。
2.二步法
(1)在含50~80mg铁蛋白的0.1mol/L磷酸缓冲液(pH7.0)中,加入稀释的戊二醛,使其最终浓度为0.05%~0.15%,总体积为1ml。
(2)置37℃作用2h后,经葡聚糖G—25 滤柱,除去未结合的戊二醛。为避免不必要的稀释,只收集脱峰的中间部分。然后加入球蛋白(铁蛋白与球蛋白之比为5:1)即铁蛋白最终浓度为15mg/ml,球蛋白3mg/ml。
(3)混合液置37℃,作用12h(无需搅拌),加入0.01mol/L 的赖氨酸以中止进一步交联。两步反应都需在0.02% NaN2防腐条件下进行。
四、电镜标本的制备方法
1.固定同常规电镜一样,应用醛类和四氧化锇双固定,以维持细胞和组织的超威结构。有文献报告以4%的甲醛溶液在pH7.2的磷酸缓冲内,在0℃进行固定,并用四氧化锇作后固定,不会影响抗原的反应能力。高锰酸钾能使大部分抗原失去活性,一般不宜采用。另外,铁蛋白标记抗体的特点之一是分子量大。因此,如用于细胞表面抗原的定位研究,可将样品直接放入固定液,否则需采取适当的措施,打破细胞膜,增强细胞对标记抗体的通透性,常采取以下方法:
(1)冻融法:将小块组织或细胞经固定后,冻融一次,使细胞膜破裂,标记抗体能进入细胞内。
(2)冰冻切片法:固定后组织快速冷冻,切成10~15μm左右薄片,溶化的切片直接浸泡于铁蛋白标记抗体液中。
(3)有报道经戊干杯固定后,浸入4×10-3mol/L洋地黄皂甙液中1~2min,能有助于增强细胞膜的穿透性,使标记抗体液进入细胞内。
2.免疫反应处理常用包埋前染色,分直接法和间接法两种。在染色前,将组织切成约10~20μm厚的薄片。
(1)直接法:将薄片直接浸泡于标记抗体液中,室温或37℃作用1~2h或更长;缓冲液(有人提倡用冷缓冲液)浸漂,除去未结合的标记抗体溶液,然后转入常规双固定和电镜包埋。
(2)间接法:
①将组织切片浸于第一抗体液中,室温30~60min。
②以大量冷缓冲液充分搅拌洗涤,至少3次,每次30min。
③浸入铁蛋白标记抗体液中,室温30min。
④如②,用缓冲液充分洗涤后,可以自然沉淀或用离心方法捞取组织片。
⑤将离心沉淀小块,用四氧化锇固定30min。
⑥常规电镜脱水、包埋、切片、染色和观察。
第三节 免疫酶细胞化学技术
一、基本原理
免疫酶细胞化学技术是以酶作为抗原抗体反应的标记物,在既不改变抗原抗体的免疫反应特异性也不影响酶活性的条件下,与相应的酶底物作用,形成一种不溶性的反应产物。在光学显微镜下观察时,要求反应的终末产物是不溶性的有色物质,具有可观察性。在电镜下观察时,则要求底物的终末产物具有较高的电子密度。由于辣根过氧化物酶(Horse Radish Peroxidase, HRP)具有稳定性强和反应特异性高等优点,是目前应用最多的酶标记物。实验方法包括酶标记抗体法、非标记抗体酶法和非标记的过氧化物酶—抗过氧化物酶技术(即PAP法,见第四章 )。
二、电镜标本制备方法
免疫酶细胞化学技术可用于包埋前和包埋后染色,但以前者应用较多。
1.单层细胞培养物免疫酶染色法
(1)用4%多聚甲醛(在0.05mol/l 磷酸缓冲液中,pH7.2)在原位固定(4℃)1h。
(2)用0.05mol/l PBS充分洗涤后,用HRP标记的抗体血清作用18h,再用PBS充分洗涤。
(3)2%戊二醛固定1h,再水洗。
(4)呈色反应,用DAB—H2O2呈色反应,水洗。
(5)1%锇酸后固定30min~1h,原位用环氧树脂包埋,光镜作半薄切片定位,作超薄切片。
2.组织切片酶标记抗体染色法
(1)1mm厚组织块,在4℃下用固定液固定后,置于含4.5%的蔗糖PBS(0.05mol/l pH7.2),4℃中漂洗,换数次固定液,过夜。
(2)随后,作10~40μm的冰冻切片,放入第一抗体血清作用12~18h,4℃,用含蔗糖的PBS洗数次。
(3)置于HRP标记抗体液(第二抗体)4℃过夜。然后用4℃含蔗糖PBS反复冲洗数次。4℃浸漂过夜。
(4)2.5%戊二醛(pH7.4磷酸缓冲液)再固定1h,4℃用含蔗糖PBS反复冲洗去戊醛,每次5min,共洗3次。
(5)DAB-H2O2显色15~30min。
(6)0.05mol/l PBS冲洗3次,每次30min,再置于室温中用1%~2%锇酸固定1h,脱水、包埋、切片复染和观察。
3.PAP包埋前染色法(Pickel 等,1975)
经固定组织、应用振动切片机(Vibratome)切35~50μm厚片,或用组织铺片,以漂浮法在反应板上进行下列步骤:
(1)正常羊血清(1:30PBS稀释),室温孵育30min。
(2)第一抗体(A种动物抗血清),4℃湿盒中48~72h。
(3)羊抗A种动物血清(1:100)室温30min。
(4)A种动物血清PAP复合物(1:100)室温30min。
(5)DAB·4HCl/H2O2(0.05%/0.01%),室温1.5min。上述每一步骤后应用PBS洗涤(5min×3次)。
(6)在解剖显微镜下检出免疫反应阳性部位,修整组织,用0.1mol/L磷酸缓冲液稀释的1%锇酸(pH7.4)后固定30min~1h。
(7)按常规电镜样品制备,脱水、包埋、超薄切片、染色观察。
4.PAP包埋后染色法(Ordronneau, 1982)
(1)将载有切片的镍网(或金网)在5%的H2O2液中蚀刻(etch)2~3min。生理盐水冲洗,滤纸吸干。
(2)正常羊血清用0.05mol/L Tris盐液(TBS)稀释成1:30,pH7.0,室温5min 。
(3)兔抗血清(第一抗体),内含1%正常羊血清,用0.05%mol/l Tris缓冲液稀释,pH7.6,4℃,48h后以Tris缓冲液洗涤。
(4)正常羊血清1:30,室温5min。
(5)羊抗兔1:10(第二抗体,0.05mol/l Tris缓冲液稀释,pH7.0),室温5min,Tris缓冲液洗涤。
(6)正常羊血清1:30,室温5min。
(7)兔PAP复合物,内含1%正常羊血清,室温3min,Tris 缓冲液洗涤。
(8)DAB—H2O2液显色(含H2O20.01%~0.03%),室温3min。
(9)4%的磷酸盐缓冲的锇酸溶液10~15min,蒸馏水洗涤(此步用于改善微细结构的反差),电镜观察。
5.PAP免疫电镜双重标记
在连续的超薄切片上进行,用包埋后染色法在相邻的两张切片上分别以不同的抗体进行免疫染色。可在超威结构水平判定细胞内抗原共存的情况。
第四节 免疫电镜胶体金标记法
金标法是Faulk和Taylor(1971)提出的,并首先用于免疫电镜。它是利用胶体金在碱性环境中带有负电的性质,使其与抗体相吸附,从而将抗体标记。当用金标记的抗体与抗原反应时,在光镜水平胶金液呈现鲜艳的樱红色,不需加外进行染色。在电镜水平,金颗粒具有很高的电子密度,清晰可辨。因此,免疫电镜胶体金标记法近年来被成功地应用于生物学的各个方面,并取得了要喜的进展,解决了一些过去未能解决的问题,80年代以来似有取代免疫电镜PAP技术的趋势。胶体金标记抗体技术在电镜水平应用有许多优点:首先,手续不如PAP法烦琐,不需用H2O2等损伤微细结构的处理步骤,对微细结构的影响较少。其次,金颗粒具有很高的电子密度,在电镜下金颗粒清晰可辨,易于与其他免疫产物相区别。因此,金标法还可以和PAP法相结合进行双重或多重染色的超微结构定位。另外,利用不同直径的金颗粒标记不同的抗体,是研究突触小泡内神经递质共存的有力工具。由于抗原抗体反应部位结合金颗粒数量的多少可进行粗略的免疫细胞化学定量研究。金标抗体还可加入培养液中,对培养细胞内抗原进行标记定位。曾有报告用金标记法于细胞内骨架的研究获满意的效果。由于金具有强烈的继发电子的能力,因此,不仅可以用于透射电镜的超薄切片观察,也可以用于扫描电镜对细胞表面的抗原、受体进行标记定位观察。金标液无毒性,对人体无损伤。胶体金及胶体金标记物的制备见第五章 第3节 。在原位分子杂交技术在电镜水平的应用中,胶体金的标记术被科技工作者认为是当前最理想的标记物(详见第二十章 )。
一、电镜水平的免疫金染色法
应用于电镜水平的免疫法,可分为包埋前染色和包埋后染色,由于包埋前染色对细胞膜的穿透性差,一般只用于细胞表面的抗原标记,如需穿透细胞膜,则需辅以冻融法或加入Triton X—100、皂素等活性剂,后者会加重细胞超威结构的破坏,因此,现较普遍采用包埋后染色,现分别介绍如下:
1.包埋后染色
(1)超薄切片厚50~70nm左右,载于200~300网孔的镍网上。
(2)置1%H2O2内10min至1h(视树脂的硬度和切片的厚度而定),以去锇酸和增进树脂穿透性,有利抗体进入。如切片很薄或于低温包埋时,此步可省略。操作时,滴入1%H2O2液1滴于蜡板上,将网的载片面轻浮于液滴上。对中枢神经系统切片,有主张以1%过碘酸钾(KIO4)代替H2O2的。
(3)双蒸水洗3次,每次10min,第1,2次洗法如(2),浮于液滴上,第3次以盛双蒸水的注射器沿镍网面冲洗,水流应有适当压力,但不宜过高强,用滤纸在网缘将水吸干。
(4)浮于正常羊血清(1:50~1:100)滴上,室温30~60min,以饱和固定剂中的游离醛基占据非特异性结合部位。
(5)PBS漂洗3min,洗1次(有人主张不洗)。
(6)滤纸吸干,孵育于第一抗体血清滴上,先室温预孵1h,再置于4℃24~36h。
(7)PBS漂洗3min,3次。
(8)PBS(内含1%的牛血清白蛋白)pH8.2中,5min,此步为胶体金结合作准备。
(9)胶体金标记抗体液1:30~1:100,淡红色为适宜稀释液,室温孵育10min至1h。
(10)双蒸水洗3min,3次。
如作双重染色,则应将镍网翻过来,用另一类抗体血清,重复上述步骤(2)~(10)。
(11)5%醋酸铀(双蒸水配制)染5min,然后用双蒸水洗。
(12)枸橼酸铀(或醋酸铅)染色5min,双蒸水洗净。
(13)电镜观察。
2.包埋前染色
(1)组织经过适当固定,为增强细胞穿透性,可在固定液中加入皂角素(Saponin),使其浓度为0.01%,经含皂角认固定剂处理5~8min后,应用0.01mol/l PBS Ph7.4冲洗12h左右,中间换洗3~4次。
(2)组织切片贴于明胶涂抹的坡片上,细胞可制成混悬液,用离心法操作或制成涂片。
(3)0.05mol/l PBS pH7.4洗3min。
(4)以1:5正常羊血清处理切片30min室温,以阻断非特异性吸附。
(5)第一抗体4℃孵育20h后室温2h或过夜。
(6)0.05mol/l TBS pH7.4洗3min×3。
(7)0.02mol/l TBS pH8.2洗3min×3,为与胶体金结合作准备。
(8)再次阻断非特异性吸附,同(4)。
(9)以金标记的第二抗体(工作浓度1:40左右)在室温下孵育1h。
(10)0.05mol/l TBS pH8.2洗3min。
(11)0.05mol/l TBS pH7.4洗3min×3
(12)1%锇酸(0.1mol/l PBS溶液)1h。
(13)双蒸水洗15min。
(14)系列酒精或丙酮脱水,包埋、超薄切片。(15)枸椽酸铅对照染色。
为增加抗非特异性染色,有的实验室倾向在TBS中加入1%小牛血清白蛋白(Bovine Serum Albumin, BSA, Sigma)。理想的免疫金染色切片,背景应清洁,无残留的金或其他无机盐颗粒,金粒集中在抗原、抗体反应部位。要获得理想的免疫金染色切片,需注意的因素很多,其中主要的如:①抗体血清的高度特异性和亲和力;②被检组织应有较高浓度的抗原;③冲洗液的清洁度,冲洗的彻底程度以及整个过程中应用的各种器皿的清洁度等;④所有溶液最好用微孔滤过器(milipore filter滤过),滤膜孔径0.2~0.45μm,所有器皿应清洁和专用。整个操作过程应在湿盒内进行,以使载网保持湿润。
二、胶体金标记蛋白A技术(Protein A-goldtechnique, PAg法)
在电镜水平应用较为广泛,因该法具有特异性中、灵敏度高、方法简便和背景染色淡等优点。蛋白A的免疫特异性在第五章 已作了介绍,PAg复合物制备方法简便,作为第二抗体,无种属特异性,可以免去不同种属动物要制备不同的特异性免疫球蛋白。PAg 复合物与包埋剂和细胞成分都极少发生非特异性的交互作用,蛋白A和金粒间非共价的结合特性既不影响蛋白A的活性,又能保持高度的稳定性,PAg复合物分子最小易于穿透组织。
1.蛋白A—金(PAg)复合物的制备(Slot 和Geuze,1981)
(1)胶体金液的制备,应用枸椽酸三钠还原法(见第五章 )。
(2)待标记蛋白质和金溶液的准备,同前。注意点是用0.2mol/l K2CO3将金溶液pH调至5.9~6.2之间。
(3)确定胶体金与蛋白A的结合用量比例。取一系列盛有0.1ml胶体金液的小玻璃管,分别加入不同量的蛋白A,5min后,再各加0.25ml 10%的NaCl。如加入的蛋白A浓度不够,不能稳住金粒,在电解质NaCl的影响下,金粒聚合沉淀,溶液由红变蓝。选择能防止溶液由红变蓝的最低浓度的蛋白A的量作为两者的结合比例。以枸椽酸钠法制成的胶体金每毫升约需要5μg蛋白A来结合,方能保证其稳定性。
(4)胶体金与蛋白A的结合和纯化,依上法测得所需的比例超过10%,即每30ml胶体中加入2mg蛋白A,5min后,加入0.3ml PEG作为稳定剂,然后以15000r/min离心45min(不同方法制备的金离心速度不同),略带红色的松散的复合物沉淀即为PAg复合物。小心弃去上清液,加入PBS冲洗,如上,松散的PAg复合物置于PBS溶液中,按0.2mg/ml的比例加入聚乙二醇作为稳定剂,保存于硅化的玻璃器皿中备用,也有主张将上述PAg复合物放入3~6ml 5%甘油—0.05%聚乙二醇—0.02%叠氮钠混合液中,再离心,弃去无色上清液后,收取管底部浓缩纯化的PAg复合物置4℃保存。
据文献报告,此PAg复合物的原液在4℃可保存达一年之久。
2.
在电镜技术的应用原则是二步标记法,可用于包埋前和包埋后染色。其主要区别于一般胶体金免疫染色在:①须1%卵白蛋白—PBs (pH7.4)或1%卵白蛋白—0.05mol/l Tris缓冲液(pH7.4)来封闭非特异性的结合部位,而不是采用羊或其它的动物的正常抗血清,因为PAg复合物能够与正常血清组中的Ig结合,从而给出假阳性结果;②在应用第一抗血清孵育和PBS冲洗后作第二抗血清即PAg复合物孵育前的准备时,应用的PBS或TBS的pH应变更为pH7.4。在变更这两步后,其它可参照本节 中包埋前、后染色法进行。也可采用下列步骤进行包埋后染色。
(1)载有超薄片的镍网或金网浮于1%卵白蛋白—PBS液滴上,室温约5min。
(2)载网不冲洗,直接移至第一抗血清液滴上,在室温孵育2h或4℃18~24h。
(3)PBS冲洗3min×2次。
(4)将PAg原液稀释10~20倍,载网浮于该液滴上,室温孵育1h。
(5)PBS冲洗5min×2次。
(6)5%醋酸铀水溶液染色,水洗。
(7)枸椽柄铅染色。
(8)电镜观察。
三、胶体金双标记技术(常用为蛋白A—胶体金)
1.单面法以不同直径的金粒分别标记两种不同的抗体,以间接法先染第一种抗体,洗净后再染第二抗体。
2.双面法以不同直径的金粒标记的抗体于镍网的两面分另进行免疫染色。本法的优点是可防止两种金标记的抗体的相互干扰,又可防止第一次应用的一抗与其相应的抗原相结合,占据了空间,第二次应用的抗体没有适合的空间使之与相应的抗原相结合。
3.异种动物抗原—抗体染色法如一抗分别用人与兔抗血清,人抗组织抗原A,兔抗组织抗原B。第二抗体分别以不同直径金粒标记的抗人和兔免疫球蛋白。由于种属不同,两种抗血清不会互相干扰,在应用一抗和二抗时都可将两种血清一次混合使用,将四步减少为两步。
4.金标记抗原检查法(Gold—labelled antigen detection method, GLAD 法)此法是Larson(1977)年首先提出的,应用放射性同位素如125I或酶标记抗原进行染色。先用特异性抗体与组织抗原反应,标记的纯抗原又与特异性抗体反应。Larson(1980,1981)又在此基础上提出应用胶体金在电镜水平进行双抗原甚至多抗原定位。GLAD原理是:双价的Ig抗体分子过量地加到有抗原的组织切片上,使分子的两个抗原结合点中有一个结合到组织抗原上,而另一端可与标记金的抗原起反应。双标记时,预先将两种不同的抗原标以不同直径的金粒,可分别与相应的第一抗体相结合,从而显示出两种抗原在组织切片的定位。此法的优点是标记物所显示出的组织抗原的部位经过两次选择,标记了抗原只能与相应的特异性抗体相结合而不能与切片中非特异性Ig相结合,因此具有较高的特异性。但由于使用此法时需备有与欲检抗原相同的纯抗原,并要对不同抗原分别进行标记,非一般实验室所能做到,所以至今尚未被广泛应用。
双面金标记法操作程序(Cai et al 1993, 改良自Bendayan et al 1982).
(1)镍网面A(树脂包埋)
①蒸馏水冲10min。
②10% H2O2蚀刻10min室温
③10%正常血清(以0.5mol/l Tris缓冲液pH7.4、含1%BSA和2%Tween20 稀释,简称TBT缓冲液)室温30min。
④以滤纸吸去多余液体,覆于特异性第一抗体1:500(Tris 缓冲液,pH7.4,含1%BSA和0.1%叠氮钠)4℃,孵育过夜
⑤彻底清洗,应用0.5mol/L Tris缓冲液pH7.2,不含BSA
⑥继之用0.5mol/L Tris缓冲液pH7.6,含1%BSA冲洗
⑦0.5mol/L Tris缓冲液pH8.2,含1%BSA,室温孵15min
⑧羊抗兔IgG标记以15nm金粒,应用0.5mol/l Tris缓冲液pH8.2、含1%BSA稀释1:40,孵育2h,室温
⑨0.5mol/L Tris缓冲液pH7.4 冲洗
⑩蒸馏水冲洗
(2)镍网面B,重复①~⑩,只在第④时更改另一特异性一抗,在⑧时羊抗兔IgG标记以5nm金粒。
(3)铀铅双重染色,电镜观察,可见二种不同直径金粒标记。
注意事项:①所有溶液均须经加有微孔滤纸(0.45μm孔,国内外现均有商品提供)的注射器过滤,过滤后直接以注射器冲洗。②滤纸最好用无纤维吸水滤纸。③冲洗在胶金标记技术上是个决定性关键,仅次于抗体血清的纯度。笔者的体会是一般应漂洗3×5min,以注射器喷水漂洗效果优于杯漂洗法,但漂洗水流需与网面平行,勿使水压破坏切片。
四、免疫电镜金—银法染色技术
关于免疫金银细胞化学的原理、试剂配制和光镜显示技术在本书第五章 已作了较详尽的叙述。免疫金银细胞化学技术说可应用于电镜水平。一般用于包埋前染色。其主要操作步骤如下:
(1)组织固定振动切片机切片10~30μm。
(2)人3%正常羊血清,含0.1%Triton X—100 的PBS孵育30min,以封闭非特异性结合部位
(3)1%硼氢化钠的PBS孵育30min。
(4)一抗37℃,2h。
(5)PBS含0.1%BSA pH7.4 冲洗3min×3次。
(6)PBS含0.1%BSA pH8.2 冲洗3min×3次。
(7)人10~15nm金标羊抗兔抗血清,工作浓度约1:10,37℃孵育45min。
(8)硝酸银液物理显影(详见第五章 第六节 )。
(9)在解剖显微镜下取免疫反应阳性部位,人1%锇酸后固定20min,常规脱水,树脂包埋。
(10)超薄切片机切0.1μm左右半薄切片,定位阳性反应部位,制超薄切片。如有暗视野微镜则更有助于定位。在暗视野前景下,金银粒呈金黄色闪光颗粒,即使微量金银也可定位,微镜则更有助于定位。在暗视野前景下,金银粒呈金黄色闪光颗粒,即使微量金银也可定位。
(11)铀—铅电镜染色,电镜观察。
免疫金银法敏感度高,金银颗粒电子密度高,反差强;应用包埋前染色可先定位阳性反应部位再作电镜超薄切片,获得阳性反应机率高,特别适用于含微量抗原的部位,如突触等。其不足是须经暗室显影,手续较烦杂,包埋前免疫染色易增加非特异性染色。另外,由于单个金粒周围结合的银粒不是固定的,受多种因素影响。因此,电镜免疫金粒染色法的金粒银粒计算不适于做半定量观察,误差较大。
第五节 其它免疫电镜技术
一、凝集素电镜标记技术
凝集素的特性及标记原理详见第五章 ,近年来,凝集素电镜标记技术应用日益广泛,且获得较为满意的效果。凝集素电镜标记技术方法较多,常用的有凝集素—酶(常用为HRP)、凝集素—生物素—酶电镜标记技术。现将凝集素—酶电镜标记技术(包埋前染色)简介如下(Sterit 和Kreatzberg,1987)。
(1)固定:常用为PLP或多聚甲醛—戊二醛固定液。如为取脑组织,可将已藻注动物在4℃过夜,次日取脑组织置0.1mol/L磷酸或二甲胂酸钠缓冲液(含7.5%蔗糖)中漂洗。
(2)振动切片机(Vibratome)切60μm的厚片。
(3)切片孵育在PBS(内含0.1mol/l CaCl2、Mgcl2和MnCl2)10min(有作者主张此步可省略)。
(4)为增强细胞通透性,切片可孵育于含0.1%胰蛋白酶和0.1CaCl2水溶液中,pH7.8,37℃孵育30min。
(5)PBS洗3次,每次2min。
(6)凝集素—HRp 1:10 在PBS中(含0.1%Triton X--100)4℃过夜,最好不断轻轻振荡。
(7)PBS洗3次,每次2min。
(8)DAB-H2O2显色。
(9)1%OSO4水溶液固定。
(10)系列酒精脱水,EPON包埋,切片。
(11)电镜沿—铀双染观察。
凝集素呈高电子密度常沉积在细胞膜上,易与电子染色相区别。
二、扫描免疫电镜技术
扫描免疫电镜技术可为研究细胞或组织表面的三维结构与抗原组成的关系提供可能性。
(一)标记物
应用于扫描电镜的标记物应能在扫描电镜可分辨的范围内,并能对细胞或组织抗原有较好的定位能力。在选择标记物时应根据研究目的而定,如标记细胞等由于体积较大,可用体积大的标记物;如鉴别阳性(标记细胞)与阴性(未标记细胞),而要定位受体等则需选用较小的,易于辨认的标记物。
常用的标记物为颗粒性标记物。依其特性可分为:
1.蛋白类 如血蓝蛋白、铁蛋白等。
2.病原体类 如烟草花叶病毒、南方菜豆花叶病毒、噬菌体T4、大肠杆菌f2、噬菌体等。
3.金属颗粒胶体金、免疫金银标记技术和同位素放射性自显影的银颗粒等。其中以金属类颗粒标记物应用最为广泛。最常用的是胶体金,胶体金商品提供的直径从3~150nm不等,扫描免疫电镜常用的金颗粒直径在30~60nm左右为宜。由于金本身系重金属,有较强的发射2次电子的作用,故不需喷镀金属膜,这是胶体金应用于免疫扫描电镜的标记优于其它标记物之处。免疫金银染色能加强细胞或组织表面金属颗粒聚集的密度。金、银粒在电镜显示为电子密度高,外形清晰的颗粒易于识别和定位。病原体标记物主要利用其特异殊的外形和结构以达到标记定位的目的,如噬菌体T4形似星形的球拍,头部大约100nm直径,呈六角形星状,尾长约100nm,由颈部与头部相接;烟草花叶病毒为15×30nm的杆状病毒,而南方菜豆花叶病毒是直径25nm的园形颗粒,这些病原体的典型外形很易于辨认。铁蛋白由于含有致密的铁离子核心具有较高的电子密度,从而达到标记定位的目的。血蛋白是由海螺类软体动物中提取的多分子聚合物,其外形为35×50nm的柱状体,多应用于病毒研究,但也有利用血蓝蛋白与过氧化物等的糖蛋白部份可与凝集素相结合的特性,进行细胞膜受体的定位。
(二)免疫标记方法
金属类标记物的免疫标记法同切片免疫染色,即将标记物与抗体相结合,通过直接或间接法显示抗原部位。胶体金可与蛋白A相结合后与IgG分子中的Fc 段相结合。哪与卵白素(a-vidin)相结合可与结合抗体的生物素(biotin)反应。免疫金银染色法在胶体金标记后,再进行银液显影。病毒(包括噬菌体)标记物多采用不标记抗体法,即搭桥法。此法的原理是采用同种动物制备抗原的特异性抗体与标记物抗体(例如兔抗A抗原与兔抗HRP)。再用另一种动物制备第一种动物血清抗体的抗体(例如羊抗兔IgG抗体)。利用后者为桥,把抗原的特异性抗体与抗标记物抗体结合起来,后者再与标记物结合,以达到定位抗原的目的,其基本原理与PAP法类同。病原体免疫标记可不用标记物显示,而利用其形态学特征定位或采用抗原抗体凝集法,其基本原理是利用病毒或病毒抗原的特异性抗体在与相应的抗原反应后,使后者之间发生交联而凝集,经浓缩后用阴性染色法(负染)便可在电镜下显示定位部位。
(三)扫描免疫电镜的具体操作步骤
1.标本处理
(1)细胞悬液:用10ml PBS 内含1mg/ml牛血清白蛋白(PBS—BSa )悬浮细胞,离心250g,5min×2。加入PBS—BSA 至105~106细胞/ml,振摇成单细胞悬液。BSA能减低生物标本的非特异性吸附,但注意浓度应适宜,过高会减弱特异性反应。
(2)细胞附着于固体支持物:由于固定与免疫标记的孵育过程会引起细胞凝集,妨碍细胞表面的暴露,而且反复的离心与悬浮会导致细胞表面形态的改变。因此,通常将悬液中的细胞粘附于过滤膜或涂有带正电荷聚合物的盖玻片上,在粘附之前可依(1)法清洗标本,以除去细胞表面的附着物。固体支持物可用涂有多聚—L—赖氨酸薄膜的载片或直径13nm、孔径0.22或0.45μm的过滤膜,载片制备方法:多聚—L—赖氨酸(Sigma)100μg/ml重蒸溶解涂抹于载片上,4℃30min后倾倒掉表面液体,令其自然干燥。注意载玻片事先需要清洁液浸泡,水漂洗过夜,然后浸泡于乙醇或丙醇中,用前取出自然干燥或用绸巾拭干。将细胞悬液(如细胞数少可事先离心,取沉淀细胞),滴于滤膜或载片上,由于多聚赖氨酸的粘附性,在固定及免疫标记过程中细胞不至于脱落。但注意勿使细胞干燥。
(3)组织切片与固体组织:组织切片如为石蜡包埋应预先脱蜡,由二甲苯经梯度酒精至水。组织切片与固体组织(勿过大)均应以PBS—BSA冲洗,并保持湿润避免干燥。
2.固定
(1)固定前用PBS—BSA冲洗5min×3。
(2)选择加入适合的固定剂;可为4%多聚甲醛+0.1%~0.5%戊二醛在pH7.4的磷酸缓冲液中。室温固定10~60min,或4℃30~120min。
(3)PBS—BSA冲洗5min×3。
(4)除去残留的自由醛基,选以下任一方法:
①0.5mg硼氢化钠/1ml PBS 10min(新鲜配制)
②0.05~0.2mol/l 甘氨醊或赖氨酸—HCl/PBs 30~60min。
③0.1~0.5mol/L氯化钠/PBs 30~60min。
④PBS—BSA冲洗5min×3。
3.免疫标记与透射免疫电镜的原则及步骤基本相同。
免疫金银染色法举例(张留保等,1997)
(1)血细胞以PBS—BSA冲洗5min×3。
(2)2%多聚甲醛—戊二醛混合液固定,4℃,1h
(3)PBS或TBS反复冲洗5min×3。
(4)胶体金免疫标记程序(略)
(5)暗室显影液显影
(6)扫描电镜样品制样
(7)观察
作者在血细胞膜外显示了密集的金银粒标记(特异性标记膜的谷胱甘肽过氧化物酶及激素肽)。
4.常规扫描电镜标本处理
(1)PBS—BSA冲洗5min×3。
(2)后固定:2.5%戊二醛0.1mol/L磷酸缓冲液,时间视样本大小而定,一般室温30min左右。
(3)PBS—BSA洗5min×3。
(4)1%四氧化锇后固定1~2h
(5)系列梯度乙醇或丙酮脱水
(6)临界点干燥或冰冻干燥
(7)喷镀碳与金
(8)扫描电镜观察
三、冷冻蚀刻免疫电镜技术
冷冻蚀刻法(Freeze Ftching),也称冷冻复型法(Freeze Replica)或冷冻切断(Freeze Fracture),是研究生物膜结构的重要方法之一。其主要步骤首先是将样品在液氮中冷冻,然后放到真空喷镀仪中切断,切断后的切面上有细胞器,其间还有冻成洋的水分。再加热使冰升华,将水份蒸发,把细胞器的膜结构暴露出来,这一步骤就称为冷冻蚀刻。如不进行蚀刻就称为冷冻切断。向暴露的膜结构上喷镀铂—碳投影,再喷碳来加固。这样就在切断的样品表面形成一层复型膜。在此复型膜上印下了细胞切面的立体结构。从真空中取出样品,把复型膜下面的组织腐蚀掉,再把复型膜捞在铜网上,在透射电镜下观察复型膜。
关于生物膜的分子结构,目前被大家公认的并为冷冻复型电镜观察所证实的是流动镶嵌型(图7-1),即脂质—球状蛋白质镶嵌模型。依照这上模型学说表明,生物膜是一种流动
图7-1 生物膜分子的流动镶嵌模型及冷冻断裂面图解
的、可塑的、镶嵌蛋白质分子的脂质双分子层的膜;脂质双分子层中每一分子分别具有两极,一端为亲水极,朝向膜的内、外表面,而另一端的疏水极朝向膜的中线部位,两排分子彼此相对,构成生物膜膜性结构的基础。蛋白质分子彼此相对有嵌入性和表在性两种,前者大约占蛋白质总量的3/4左右,外形近似球状,镶嵌在脂质分子层的不同深度内,而后者则大多附着于细胞膜的胞质面。在冷冻劈裂后,生物膜的水平断面大多发生在单位膜的疏水极。因此,膜的亲水部,分别命名为PS(与细胞质,核质或线粒体内基质相邻的面)和ES(与细胞外间隙或细胞内间隙或细胞内间隙相邻的面,如内质网腔、核质间隙、线粒体内、外膜之间的腔和其它各种泡的腔等)。膜的疏水部、亦即劈裂面分别叫做EF(面向细胞质、核质、或线粒体基质的面)和PF(向细胞外间隙或内间隙的面)。从70年代初期开始,冷冻蚀刻免疫电镜技术已开始在应用,但由于免疫标记必须在冷冻蚀刻步骤以前进行,所以仅能标记细胞外表面(ES)。80年代开始建立了断裂—标记细胞化学方法,将细胞膜劈开后,中央的两侧断面(EF与PF)以及各种细胞器的膜的各个表面及细胞质与核质都能被标记,为此技术的广泛应用创造了条件。应用此法还可对抗原与受体分子进行定量统计。
1.冷冻蚀刻表面标记免疫电镜技术
(1)新鲜或固定的细胞进行直接法或间接法免疫标记。
(2)PBS(pH7.5)冲洗3min×2,加入1mmol/l MgCl2蒸馏水洗洗3min×3,离心沉集细胞。
(3)将细胞团置于小纸板上,入液氮冷却的Freon中,取出入冷冻蚀刻仪中进行断裂操作,再于-100℃蚀刻1min 。
(4)制做断裂面复型。
(5)再次氯酸钠清洗复型,蒸馏水洗后进行观察。
本法可显示断裂暴露的PF位于中央,周围则是蚀刻后露出来的ES,标记物只出现在ES上。
2.断裂—标记免疫电镜技术
此法是先进行冷冻断裂,再做免疫标记,从而可以对断裂开的各种膜结构及胞浆断面进行标记。
(1)临界点干燥法
①固定:1.0%~2.5%戊二醛PBS液4℃1~2h,PBS冲洗3min×3。
如为细胞悬液,可加入30%BSA后加入1%戊二醛,使BSA凝胶化,将凝胶切成2mm左右的小块,用30%的甘油—PBS浸透后置于用液氮冷却的Freon中冷却。
②冷冻断裂,将冰冻的凝胶小块放在盛有液氮的培养皿中,培养皿放置于二氧化碳—液氮槽中,用预冷的解剖刀切割凝胶小块进行冷冻断裂。
③解冻,置碎块于30%甘油—1%戊二醛磷酸缓冲液中解冻。
④置换甘油,放入1mmol/l 氨基乙酰甘氨酸磷酸液去甘油,PBS冲洗,3min×2。
⑤免疫标记。
⑥1%锇酸,室温固定30min。
⑦系列梯度乙醇脱水,临界点干燥,喷镀铂—碳膜,次氯酸钠清洗复型,蒸馏水洗,捞于有Formvar 膜铜网上透射电镜观察。
(2)超薄切片法
步骤:①至⑤同临界点干燥法。
⑥1%锇酸,室温固定2h,系列酒精或丙酮脱水,常规电镜包埋。
⑦切半薄片,光镜定位合适的断裂部位,再切超薄切片,铀铅染色,透射电镜观察。
断裂标记法目前文献报告应用较多的是植物凝集素—胶体金免疫标记技术,常用的如刀豆球蛋白(Con A)-- 胶体金免疫标记技术,如第六章 所述,Con A 能与细胞膜中的甘露糖结合,能标记内质网膜、核被膜以及细胞膜的EF面,有助于糖蛋白在超微结构水平的定位。
为保证实验结果的准确性,每组实验在免疫标记阶段应设立对照组。对照组的设计同第一章 总论中的免疫对照染色。
参考文献
1.Altaman IG,et al.Rapid embedding of tissue LowicrylK4m for immunoelectron microscopy.J Histochem Cytochem, 1984,32:1217
2.Bendayan M.Double immunocytochemical applyingthe protein A-Gold techniqnes.J Histochem Cytochem, 1982,30:81~85
3.Cai WQ(蔡文琴),et al.Colocalization of vasoactivesubstances in endothelial cells of human umbilical vessel.Cell Tissue Res, 1993,274:533~538
4.De Mey J.Clloidal gold probes inimmunocytochemistry.In :Immunocytochemistry(Eds:Polak JH and Noordren SV)1983;82,wright PSG Bristol LondonBoston
5.Faulk WP and Taylor GM, Am immunocolloid method for theelectron microscope.Immunocytochemistry, 1971,8:1081
6.Mclean I and Nakane PK.Periodase-Lysine-paraformaldehyxefixative for immunoelectron microscopy.J Histochem Cytochem, 1974,22:1077
7.Ordronneaa P.Technique inimmunocytochemistry(Eds:Bullock GR and Petrwoz P, 1982, 269~280
8.Roth J, et al.Application of the protein A-goldtechnique for electron microscopic demonstration of hormone.Endocrinology, 1981,108:247
9.Spour RC and Morlarty GC.Improvements of glycolMethacrylats.I.Its as an embodding medium for electron microscopic studies.J His-tochem Cytochem, 1977,25:163
10.Sterit WJ and Kreatzberg GW.Lectin binding by resting andreactive microglia.Neurocytol, 1986,16:249
11.Solt JW and Geuze HJ.A new method of preparing goldprobes for multiple-labelling.Cytochemistry.Eur J Cell Biol, 1985,38(1):87
12 .Tokuyasu KT.Immunocytochemistry on ultrathinfrozen sections.Histochem J, 1980,12:381
13.Vacca LL, et al.A modified peroxidase procedurefor improved localization of tissue ontogens,localization of substance Pin ratspinal cord.J Histochem Cytochem, 1980,28:297~307
14.Wang Bao –Le, et al.Simplified purification andtesting of colloidal gold probes.Histochemistry, 1985,83:109~115
15.Cai WQ(蔡文琴),et al.Localization of neuropeptide Y andatrial natriuretic petide in the endothelial cells of human umbilical blood ves-sels.Cell Tissue Res, 1993,272:175~181
16.蔡文琴.胶体金标记法在免疫细胞化学电镜的应用.解剖学杂志,1985,8(2):19
17.张承志.软型包埋剂GMA制作薄切片的原理及其应用.解剖学杂志,1986,9(1):73
18.李文镇主编.组织细胞冷冻复型图谱.人民卫生出版社,1981
19.孙榆,蔡文琴,李巍,等.改良低温包埋胶体金免疫电镜技术.第三军医大学学报,1991,13(8):417~472
第八章 蛋白质与多肽激素的放射免疫分析
第一节 概述
1960年,美国学者Yalow 和Berson 创立了放射免疫分析(Radioimmunoassay,RIA),并首先用于糖尿病人血浆中胰岛素含量的测定。这是医学和生物学领域中方法学的一项重大突破,开辟了医学检测史上的一个新纪元。它使得那些原先认为是无法测定的极微量而又具有重要生物学意义的物质得以精确定量,从而为进一步揭开生命奥秘打开了一条新的道路,使人们有可能在分子水平上重新认识某些生命现象的生化生理基础。其后30年中,内分泌科学的飞速进展,充分证明了这一超威量分析技术的巨大推动力。1977年,这项技术的发明者荣获诺贝尔生物医学奖。随后这一崭新的技术迅速渗透到医学科学的其它领域,如病毒学、药理学、血液学、免疫学、法医学、肿瘤学等,以及与医学生物学相关的学科,如农业科学、生态学及环境科学等。放射免疫分析的物质,由激素扩大到几乎一切生物活性物质。我们放射免疫分析研究起步于1962年,并迅速发展与普及,对我国生物医学的进展起着很大的促进作用。
一、放射免疫分析的优缺点
(一)RIA的优点
放射免疫分析具有许多其它分析方法无可比拟的优点。它既具有免疫反应的高特异性,又具有放射性测量的高灵敏度,因此能精确测定各种具有免疫活性的极微量的物质。
1.灵敏度高一般化学分析法的检出极限为10-3~10-6g,而RIA通常为10-9(毫微克,ng)、10-12g(微微克,pg),甚至10-15g(毫微微克,fg)、10-18g(微微微克,ag)。
2.特异性强由于抗原—抗体免疫反应专一性强,所被测物一定是相应的抗原。良好的特异性抗体,能识别化学结构上非常相似的物质,甚至能识别立体异构体。
3.应用范围广据不完全统计,目前至少已有300多种生物活性物质已建立了RIA。它几乎能应用于所有激素的分析(包括多肽类和固醇类激素),还能用于各种蛋白质、肿瘤抗原、病毒抗原、细菌抗原、寄生虫抗原以及一些小分子物质(如环型核苷酸等)和药物(如地高辛、毛地黄甙等)的分析,应用范围还在不断扩展。近年来由于小分子半抗原制备抗体的技术有很大的发展,有人预测几乎所有的生物活性物质,只要其含量不低于RIA的探测极限,都可建立适当的RIA法。
4.操作简便RIA所需试剂品种不多,可制成配套试剂盒;加样程序简单一次能分析大量标本,标本用量也少;反应时间不长;测量和数据处理易于实现自动化;RIA属体外分析技术,对患者无任何辐射危害。
(二)RIA的缺点
1.只能以免疫反应测得具有免疫活性的物质,对具有生物活性百失去免疫活性的物质是测不出的。因此RIA结果与生物测定结果可能不一致。
2.由于使用了生物试剂,其稳定性受多种因素影响,需要有一整套质量控制措施来确保结果的可靠性。
3.灵敏度受方法本身工作原理的限制,对体内某些含量特别低的物质尚不能测定。
4.由于放射免疫分析是竞争性的反应,被测物和标准物都不能全部参与反应,测得的值是相对量而非绝对量。
5.存在放射线辐射和污染等问题。
尽管RIA存在以上缺点,但它毕竟是定量分析方法的先进技术。随着科学技术的进步,放射免疫分析技术将会得到更加广泛、更加深入的发展。
二、基本原理
放射免疫分析技术,是把放射性同位素测定与抗原、抗体间的免疫化学反应两种方法巧妙地结合起来所形成的一种超威量物质的测定方法。
RIA的基本原理,是利用标记抗原(*Ag)和非标记抗原(Ag)对特异性抗体(Ab)发生竞争性结合。竞争结合反应可用下式表示:

在上述反应系统中,当只有*Ag和Ab时,只产生*Ag—Ab复合物,并保持可逆的动态平衡。如反应系统中同时加入Ag,因Ag 与*Ag 免疫活性完全相同,故与Ab具有相同的亲合力。当*Ag为一定量、Ab为有限量、Ag 与* Ag 的量之和超过Ab上的有效结合位点时,*Ag –Ab复合物的生成量与Ag 的量之间呈一定的函数关系。即当Ag 量少时,Ag –Ab生成量多,而*Ag –Ab生成量增多,游离的*Ag 减少。可见*Ag –Ab复合物生成量是受Ag含量制约的。因此,在放射免疫分析中,用已知不同浓度的标准物和一定量的*Ag及限量的Ab反应,采取一定方法将B与F分开,即可算出该标准物在各浓度下*Ag –Ab复合物的结合百分率(B/T)。
这一反应过程,可用以下简图(图8-1)说明。图中黑圈表示标记抗原,白圈表示非标记抗原,长条代表抗体,每个抗体有两个结合位点,标记抗原与非标记抗原对抗体有同等的结合能力。
图8-1 放射免疫分析原理示意图
从图中可见,当标记抗原与抗体量一定时,结合率(B/B+F)随抗原量增加而降低。在实际工作中,以B/T的值为纵座标,标准物的浓度为横座标,绘成曲线,即竞争性抑制曲线,或称准确曲线。将未知浓度的样品按同样条件操作,所得结合率(%)与标准曲线相比,即可查出样品中待测抗原的浓度。
放射免疫分析典型的操作程序如下:首先配制一系列已知浓度的标准溶液,并各取一定体积;于其中加入一定量的标记抗原和特异性抗体;在一定条件下使之反应平衡后,采取适当方法将B与F分离;分别测量其放射性;绘制标准曲线(图8-2)。对样品中抗原的测定,则可在同样条件下操作,在标准曲线上查得含量。
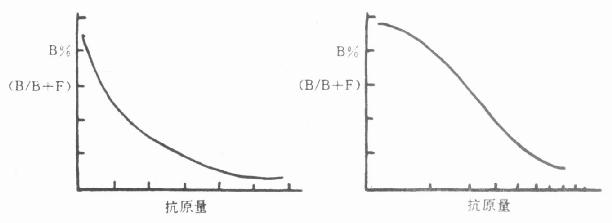
图8-2 标准曲线
左:横座标为等份刻度; 右:横座标为对数刻度
由此可见,要成功地进行放射免疫分析,必须解决好以下3个关键性技术问题:
(1)标记抗原:要求其纯度高、免疫化学活性好、比放射性强、用量适当。
(2)制备抗体:要求其特异性高、选用的稀释度适当。
(3)分离B与F:理想的分离方法应当是分离完全、稳定可靠、操作简单、适用范围广。
第二节 放射性碘标记
在RIA中,标记抗原质量的优劣,直接影响测定结果,必须制备比放射性强、纯度高的标记抗原,并保持免疫活性不受丧失。
一、同位素的选择
同位素有稳定性和放射性两种。放射性同位素可利用其衰变时放出的放射线进行测量,这种测量较灵敏而方便,故多用放射性同位素。标记抗原,常用的放射性同位素有3H、14C、131I和125I等。在使用上各有其优缺点,可根据所进行的放射免疫分析的类型特点,标记物制备和供应情况以及实验室设备条件等作适当的选择(表8-1)。大多数抗原分子中都含有C、H等原子,所以用14C或3H标记不改变抗原的结构及其免疫学活性,且14C、3H半衰期长,所标记的抗原长时间放置后仍可使用,这都是其优点。14C或3H标记的不足之处是操作较繁琐,并难以获得高比放射性的标记物;3H及14C放出的都是弱β射线,需用较昂贵的液体闪烁计数器方能获得较高效率的测量,且测定操作也较麻烦。但某些抗原用放射性碘标记容易丧失免疫化学或生物学活性者,则仍以采用3H或14C标记物为佳。
表8-1 标记抗原常用的放射性同位素及其性质
| 放射性元素 | 半衰期 | 射线种类及能量(百万电子伏特) | |
| β | γ | ||
| 14C | 5720年 | 0.155 | - |
| 3H | 12.5年 | 0.0189 | - |
| 125I | 60天 | - | 0.035 |
| 131I | 8.05天 | 0.608,0.335,0.250 | 0.364,0.637,0.722 |
大多数抗原分子中是不含碘的,引入碘原子就改变了抗原的分子结构,往往容易损伤抗原的免疫化学活性;且放射性碘的半衰期较短,标记物放置后因衰变使放射性降低,因而需要经常制备标记物或要求能定期提供放射性碘标记都能适用,放射性碘放出γ—射线,用一般晶体闪烁计数器就能获得较高效率而精确的测量,测量操作也很简单。由于这些突出的优点,目前在放射免疫分析中,使用放射性碘标记物最多。
从应用角度来看,131I和125I又各有其优缺点,可根据实验的要求、仪器的条件和放射性碘制剂的规格等条件合理选用。但相对而言,125I有较多的优点,一是半衰期适中,允许标记化合物的商品化及贮存应用一段时间;二是它只发射28keV能量的X射线和35keV能量的γ射线,而无β粒子,因而辐射自分解少,标记化合物有足够的稳定性。放射性碘适用于放射免疫分析许多对象(包括蛋白质、肽类、固醇类、核酸类以及环型核苷酸衍生物等)的标记,且操作简单,一般实验室都不难做到。
二、蛋白质与多肽激素的放射性碘标记
要制备高比度、高纯度与免疫化学活性好的标记物,首先要有高纯度、良好免疫活性的抗原。用作放射标记加网免疫分析的特异性,所以若用纯度不高的抗原作标记,则标记后必须采取适当的步骤除去杂质,以获得高纯度的标记物。标记对象的纯化应尽量采用温和的方法,否则在纯化操作中已受潜在性损伤的蛋白质(这时表面上活性可能还是良好的),再经标记反应时所受的损伤,活性就会显着降低,影响以后的放射分析结果。有了好的纯抗原,还要采用适当方法加以标记,尽量获取高比放射性、而又能保持良好的特异免疫化学活性的标记物。这些都是放射免疫分析能取得高特异性和高灵敏度的关键问题。
多肽激素与蛋白质多用碘标记,最常用的是125I。碘化反应的基本过程如下:通过氧化剂的作用,使碘化物(125I-)氧化成的碘分子(125I2),再与多肽激素、蛋白质分子中的酪氨酸残基发生碘化作用。所以不管采用哪一种放射性碘标记法,标记的化合物内部必须有碘原子可结合的基团,即结构上要含有酪胺基或组织胺残基。凡蛋白质、肽类等抗原,在结构上含有上述基团的可直接用放射性碘进行标记。如不含上述基团的,放射性碘无法标记,必须在这些化合物的结构上连接上述基团后才能进行碘标记。
因此影响蛋白质、多肽碘化效率的因素,主要决定于蛋白质、多肽分子中酪氨酸残基的数量及它们在分子结构中暴露的程度;此外,碘化物的用量、反应条件(pH、温度、反应时间等)及所用氧化剂的性质等也有影响。
常用的标记方法有:
(一)氯胺T法
氯胺T法标记效率高、重复性好、试剂便宜易得,是目前使用最多的碘标记方法。
1.原理 氯氨—T(Chloramine--T)是一种温和的氧化剂,在水溶液中产生次氯酸,可使碘阴离子氧化成碘分子。这活性碘可取代肽链上酪氨酸苯环上羟基位的一个或两个氢,使之成为含有放射性碘化酪氨酸的多肽链。
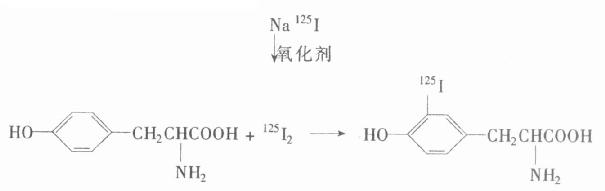
2.方法 以125I—AVP的制备为例。
(1)碘化反应:AVP5μg+0.5mol/lPB50μl(pH7.5)+1251800μCi,混合后,加入新配置的Ch—t 30μg/15μl(0.05mol/l PB, pH7.5)。迅速振荡混匀,室温下反应40s。
(2)终止碘化反应:加入还原剂偏重亚硫酸钠40μg/20μl(0.05mol/l PB, pH7.5),以终止碘化反应。
(3)Bio—Gel P2层析纯化:将碘化反应混合液注入Bio—Gel P2柱,用0.1n HAC溶液洗脱,分部收集,每2min收集一管,共收集60管。
(4)放射性测量:测定各收集管的放射性,出现两个峰,第一峰为125I—AVP,第二峰为游离碘盐峰。第一个峰中计算最高的几管,留下备用。
为了解标记抗原的质量,每次碘标记后应计算出碘的利用率,标记上多少放射性碘,以及每微克抗原结合上多少放射性碘。
(5)标记抗原的贮存:经纯化与检查后的标记物、加入1/8体积的异丙醇,分成若干小份,置于铅罐中,在-20℃以下的冰箱中贮存备用,应避免反复冻融。标记抗原在贮存中是不稳定的,这是因为:一是脱碘,标记的碘从原来位置上脱落,变成游离碘;二是蛋白损伤、变性,成为聚合大分子或断键成小分子碎片。由于上述原因,使B/F明显降低,标准曲线斜率变小,以致不能使用,故需分离纯化,其方法是用SephadexG100长柱(40~80cm )过柱,洗脱后出现3个峰。第1个峰分子量大,是蛋白变性的聚合的大分子,尚保留部分抗原决定簇,免疫活性弱;第2个峰是纯抗原的蛋白峰,免疫活性好;第3个峰是游离125I或小分子碎片,不具备免疫活性。收集到的第2个纯抗原蛋白峰,免疫活性好;第3个峰是游离125I或小分子碎片,不具备免疫活性。收集到的第2个纯抗原蛋白峰,其性能类似于新鲜标记的抗原。分离纯化的方法解决了标记抗原的贮存、长期使用问题,特别对来之不易的抗原更显得重要。
2.注意事项
(1)放射性碘源的选用:无载体的131I或125I均可用于碘化标记,但应尽量选用新鲜的、比放射强度高的、含还原剂量少的放射性碘源。碘源的比放射强度最好≥50~100mCi/ml,至少也要>30mCi/ml,否则加入碘源的容量要增加,随着带入碘源中含有的还原剂(为放射性碘源运输保存所需加入)量也增加,这将会显著降低碘利用率及标记蛋白比放射强度。放置较久和放射性碘源,一方面因衰变致比放射强度降低,另一方面因水的辐射化学产物增多(主要是131I源),都会降低标记时的碘利用率。放射性碘源含还原剂(如Na2S2O5等)量多时,会抵消氯胺T的作用,降低碘利用率,甚至导致标记完全失败。放射性碘源要用无载体的,标记所用全部用具和试剂必须不含碘;只要有极少量的碘的污染,非放射性碘就会稀释放射性碘,使放射性碘利用率显著降低。为了便于放射性防护和除污染,以及减少射线对蛋白质分子的损伤,标记投入的放射碘量不宜过大,一般以4MFOPS(每秒兆浮点处理指令)。在各种图像输入设备中,以视频摄像机为图像的基本输入设备,也可以配接视频录像机、数字扫描仪和连接扫描电镜的专用接口。高分辩彩色监示器为图像的基本输出设备。该系统还具备有自动移位的载物台和自动聚集控制系统可供用户选择。MIAS系列图像分析仪的软件系统是在DOS(磁盘操作系统)3.2以上环境开发的,全部采用C语言和汇编语言(80286宏汇编,TMS340系列专用汇编)进行编程,共98个功能模块。主要功能包括初始化及环境设置、图像输入和输出、图像编辑、图像增强、图像分割、图像测量、实用程序等。测量参数包括一维和二维目标的各种形态参数,灰度参数和纹理参数等数十种。该系统在长期为华西医科大学、第一和第四军医大学、北京军事医学科学院、上海医科大学、湖南医学院、哈尔滨医科大学等数十个医学单位的服务中,积累了许多有效的实用算法和功能、用户界面和处理速度方面已达到和超过某些进口中高档图像仪的水平。
(二)国外情况
英国的主要生产厂家有剑桥仪器公司(Cambridge Instruments Company),在图像的测量、计算、比较、分类等功能上都比过去的产品更完善,使用了大功率的新型计算机,使用QUIPS程序可获得各种图像分析测量的数据,重现性好。操作者不必精通程序设计语言,只需接受几小时使用QUIPS语言的教学训练即可。
英国Ealing—Beck公司生产Histotrak图像分析系统,如用透射光,该仪器能自动改变放大倍数。具有阴影校正系统。最使研究工作者感兴趣的是该仪器中设有坐标发生器,能使被检图像的坐标数据通过台式计算机在输出打印机上打印出来。
英国Micro Measurements 公司生产电视扫描图像分析仪,成本较低,用途较多。
英国Joycez—Loebl公司生产Magiscam图像分析系统,操作较简单,可解决一般图像分析问题,并可进行彩色图像分析,在医学和放射学等方面适用。
美国的主要厂家有美国美敦设备公司(Milton Roy Company),生产OMNICON图像分析仪,据上海多国仪器仪表展览会资料介绍,Ommicon图像分析系统有Omnicon Alpha500用于常规检验和研究。Ommicon FAS—III 用于医学和生物学领域较合适。Omnicon 3500为一种多用途的系统,配有64K通用NOVA4×计算机。Omnicon 5000是该公司发展的使用起来最方便的定量图像分析仪。Omnicon 7500是为了对图像作实时分析而设计的产品。
德国Leitz公司制造Leitz T.A.S图像分析仪,图像输入装置是一台Leitz Orthplan显微镜,该仪器还包括一台TU—58双盒式磁带机,可用于存贮程序、数据、图像。该公司产品的新型号是Leitz T.A.S PLUS,增加了自动调焦、自动图像旋转等功能。
德国Carl Zeiss公司的产品是Micro—Videomat,主要优点是有适用于计算机和大型计算机的程序。型号有Micro Videomat 2型、3型等。
德国的OPTON公司生产IBAS2000图像分析系统,可应用于显微图像分析的所有领域,如放射自显影、染色体分析、真色图像信息、细胞学分析、用连续切片来重建原形、颗粒大小分析、纤维分析、可测定交叉的或重叠的纤维长度及纤维的长/宽比例等。
德国VEB Zeiss Jena公司生产EPIQUANT图像分析仪,可进行自动或半自动分析。
日本浜板电视公司生产一系列特殊用途的、测量特殊参数的自动分析系统,如其中有一种称为Multianalyser的,与显微镜相结合,可用来测面积、长度等。
对国内外资料收集比较局限,总之购置仪器时要充分了解性能、对价格作比较、才能获得满意的结果。
参考文献
1.Jenkinson G. An introduction tothe operation and capabilities of image analysis systems.International Labmate,1987:12(3~4)
2.WeibelER. Stereological methods –practical methods for biological morphometry. London:Academic Press,1979
3.McMillanPJ. Objective evaluation of immunohistochemically stainned tissues using animage array processor. 6h International congress for Stereology, 1983
4.AaltoML, et al . Morphometric approach to immunohistochemisiry:carcinoembryonic antigen(CEA)inovarian tumours. Acta Stereologica, 1982;1(2):347~356
5.BoudierJA. et al. Stereological analysis of the rat neurohypophysis. ActaStereologica, 1982;1(2):323~328
6.StewardMG, et al. Stereologlcal analysis of synapses in the brain of the domesticchick following avoidance learning. 6th International Congress for Stereology,1983
7.CambridgeInstruments Issue 2, 1987
8.WiernikG, et al. A quantitative comparison between normol and carcinomatous squamousepithelia of the uterine cervix, British Journal of Cancer, 1973;28:488
9.杨光,等.豚鼠腹腔神经节 小强荧光细胞超微结构的定量分析.中国第四届体视学与图像分析学术会议论文汇编(第一集),1987]
10. 孙培懋,等译.图像分析入门.北京:计量出版社,1983
11. 管汀鹭译.显微术中的分析与定量方法.北京:科学出版社,1983
12. 杨正伟.生物组织的形态计量研究中应特别注意的两个问题:参照空间的“陷井”和样本含量问题.川北医学院学报,1992;7(3):73~76
13. Crus- Orive LM, et al:Recent stereological methods for cell biology:abrief survey. Am J Physiol, 1990;258(Lung Cell/Mol . Physiol. 2 ):L148~L156
14.Baddeley AJ, et al.:Estimation of surface area from vertical sections. J of Microscopy,1986;142:259~276
15.Mattfeldt T, et al. Orthogonal triplet probes:an efficient method for unbiased estimationof length and surface of objects with unknown ori-entation in space. J of Microscopy, 1985;139(3):279~289
第十章 流式细胞术(FCM)在免疫细胞化学中的应用
近些年来,流式细胞分析技术已经成为细胞生物学、肿瘤学、免疫学等基础和临床医学领域中的一项新的有效工具。本章 主要介绍流式细胞分离及分析的基石原理、流式细胞计的基本结构和操作、流式细胞术在免疫细胞化学中应用的技术思路、途径和方法,以及它们在临床医学中的应用。对FCM在外周血白细胞的免疫组织化学分析方面的应用作重点介绍。
第一节 流式细胞术简介
一、流式细胞术发展简史
流式细胞术(Flow Cytometry, FCM)是一种可以对细胞或亚细胞结构进行快速测量的新型分析技术和分选技术。其特点是:①测量速度快,最快可在1秒种内计测数万个细胞;②可进行多参数测量,可以对同一个细胞做有关物理、化学特性的多参数测量,并具有明显的统计学意义;③是一门综合性的高科技方法,它综合了激光技术、计算机技术、流体力学、细胞化学、图像技术等从多领域的知识和成果;④既是细胞分析技术,又是精确的分选技术。
概要说来,流式细胞术主要包括了样品的液流技术、细胞的分选和计数技术,以及数据的采集和分析技术等。FCM目前发展的水平凝聚了半个世纪以来人们在这方面的心血和成果。
1934年,Moldavan1首次提出了使悬浮的单个血红细胞等流过玻璃毛细管,在亮视野下用显微镜进行计数,并用光电记录装置计测的设想,在此之前,人们还习惯于测量静止的细胞,因为要使单个细胞顺次流过狭窄管道容易造成较大的细胞和细胞团块的淤阻。1953年Crosland –Taylor根据雷诺对牛顿流体在圆形管中流动规律的研究认识到:管中轴线流过的鞘液流速越快,载物通过的能力越强,并具有较强的流体动力聚集作用。于是设计了一个流动室,使待分析的细胞悬浮液都集聚在圆管轴线附近流过,外层包围着鞘液;细胞悬浮液和鞘液都在作层液。这就奠定了现代流式细胞术中的液流技术基础。
1956年,Coulter在多年研究的基础上利用Coulter效应生产了Coulter 计数器。其基本原理是:使细胞通过一个小孔,只在细胞与悬浮的介质之间存在着导电性上的差异,便会影响小孔道的电阻特性,从而形成电脉冲信号,测量电脉冲的强度和个数则可获得有关细胞大小和数目方面的信息。1967年Holm等设计了通过汞弧光灯激发荧光染色的细胞,再由光电检测设备计数的装置。1973年Steinkamp设计了一种利用激光激发双色荧光色素标记的细胞,既能分析计数,又能进行细胞分选的装置。这样就基本完成了现代FCM计数技术的主要历程。
现代的FCM数据采集和分析技术是从组织化学发源的,其开拓者是Kamentsky。1965年,Kamentsky在组织化学的基础上提出了两个新设想:(1)细胞的组分是可以用光光度学来定量测定的,即分光光度术可以定量地获得有关细胞组织化学的重要信息。(2)细胞的不同组分可以同时进行多参数测量,从而可以对细胞进行分类。换句话说,对同一细胞可以同时获得有关不同组分的多方面信息,用作鉴别细胞的依据。Kamentsky不仅思路敏捷,而且能身体力行。他是第一个把计算机接口接到仪器上并记录分析了多参数数据的人,也是第一个采用了二维直方图来显示和分析多参数的人。
流式细胞术在细胞化学中的应用的先驱者是Van Dilla和美国的Los Alamos小组。他们在1967年研制出流液束、照明光轴、检测系统光轴三者相互正交的流式细胞计的基础上,首次用荧光Feulgen反应对DNA染色显示出DNA的活性与荧光之间存在着线性关系,并在DNA的直方图上清楚地显示出细胞周期的各个时相。Gohde 和Dittrich接着把这项技术推向实用,他们用流式细胞术测定细胞周期借以研究细胞药代动力学问题。FCM用于免疫组织化学中的关键是对细胞进行免疫荧光染色,其它和在细胞化学的应用并没有多大差异。
近20年来,国内外在FCM上都做了不少的研究和应用工作,也取得了不少成果。特别是随着仪器和方法和日臻完善,人们越来越致力于样品制备、细胞标记、软件开发等方面的工作以扩大FCM的应用领域和使用效果。FCM在免疫组织化学中的应用也大致差不多,并注重了在临床应用的推广。
二、流式细胞计的基本结构和工作原理
流式细胞计是对细胞进行自动分析和分选的装置。它可以快速测量、存贮、显示悬浮在液体中的分散细胞的一系列重要的生物物理、生物化学方面的特征参量,并可以根据预选的参量范围把指定的细胞亚群从中分选出来。多数流式细胞计是一种零分辨率的仪器,它只能测量一个细胞的诸如总核酸量,总蛋白量等指标,而不能鉴别和测出某一特定部位的核酸或蛋白的多少。也就是说,它的细节 分辨率为零。国外又把流式细胞计称作荧光激活细胞分选器(Flu-orescence Activated Cell Sorter, FACS)。美国Becton—Dickinson 公司生产的流式细胞计系列均冠以FACS字头。目前我国国内使用的仪器多为美国、西欧及日本等国的产品,国内有些单位也已研制成功,但尚无定型产品面市。
1.流式细胞计的基本结构流式细胞计主要由四部分组成。它们是:流动室和液流系统;激光源和光学系统;光电管和检测系统;计算机和分析系统。图10-1为其结构示意图。
图10-1 流式细胞计结构示意图
(1)流动室和液流系统:流动室由样品管、鞘液管和喷嘴等组成,常用光学玻璃、石英等透明、稳定的材料制作。设计和制作均很精细,是液流系统的心脏。样品管贮放样品,单个细胞悬液在液流压力作用下从样品管射出;鞘液由鞘液管从四周流向喷孔,包围在样品外周后从喷嘴射出。为了保证液流是稳液,一般限制液流速度υ90%
(2)假阳性:较低,SLE病人亲属和配偶,密切接触者可阳性,常为IgM。
(3)假阴性:病程四个月以内者及治疗后(多为青年SLE病人)可阴性。
3.临床用途
(1)鉴别SLE和DLE(盘状红斑狼疮):DLE和LBT阴性。
(2)鉴别SLE和其它ANA阳性疾病:其它ANA阳性疾病LBT常阴性。
(3)有助于判断病情和预后:病情严重者不仅沉积的Ig类别多,而且荧光强,有IgG、IgA沉积者比仅有IgM沉积者病情重,Ig类别和荧光强度与肾病的关系尤为明显。
(4)可能有助于估价疗效。
第六节 免疫细胞化学技术在真皮纤维化性疾病研究中的应用
真皮结缔组织由数量不多的细胞和丰富的细胞外基质组成。细胞成分包括成纤维细胞、肥大细胞、巨噬细胞和少量淋巴细胞等。细胞外基质包括:①胶原:真皮中胶原纤维由I型和II型胶原构成,成熟真皮中I型胶原约占胶原的85%,越年幼的皮肤含III型胶原的比例越高。②弹性蛋白。③非胶原糖蛋白:如纤维粘连蛋白。④蛋白聚糖:为氨基聚糖(亦称糖胺聚糖)与核心蛋白质构成的共价化合物。其中氨基聚糖可分为透明质酸、硫酸软骨素、硫酸皮肤素、硫酸角质素、肝素和硫酸乙酰肝素等。
真皮纤维化是以胶原为主的细胞外基质成分在真皮中过度积聚的结果。它既见于硬皮病等皮肤病中,还见于皮肤创伤愈合过程中发生的肥厚性瘢痕和瘢痕疙瘩中。现将免疫细胞化学技术在上述常见的真皮纤维化性疾病研究中的应用简介如下:
一、确定细胞外基质成分在真皮的分布
利用各种细胞外基质的抗体,借助免疫细胞化学技术对细胞外基质在真皮中的分布进行定位,尚可经图像分析系统对染色强度等参数定量化,所获得数据经统计学处理能更准确地反映细胞外基质分布和含量上的量化差异程度。用ABC法显示,硬皮病真皮中III型胶原大量沉着,其分布方式与免疫电镜所见相符,即标记III型胶原抗体的细纤维大量聚积于真表皮交界处基底膜下;而在真皮深部,这种细纤维与标记I型胶原抗体的粗纤维相间分布,形成光镜下所见的粗大纤维束。纤维粘连蛋白也可大量沉积于硬皮病真皮深层胶原纤维束之间,但有些文献报道的结果并不一致。免疫荧光法显示,纤维粘连蛋白在肥厚性瘢痕和瘢痕疙瘩中量沉积,呈线状或卷曲排列,其分布符合组织学上所见结节 性结构的特点。近来用不同蛋白聚糖的单克隆抗体,借助APAAP法发现,肥厚性瘢痕组织中含有大量4-或6-硫酸软骨素,只有少量硫酸皮肤素。其中血管周围浸润的T淋巴细胞与4-硫酸软髓素密切相关。
二、研究纤维化皮损中细胞的表型特征和(或)功能状态
(一)成纤维细胞
1.合成功能活跃除了上述对已形成胶原的分布进行观察外,利用抗体I型前胶原氨基末端的单克隆抗体,可确定新合成未加工的I型前胶原。正常人皮肤显示了真表皮交界部I型前胶原的灶性沉着。而硬皮病皮损及其未受累皮肤中,真皮上部有连续广泛的I型胶原着色。这反映了硬皮病成纤维细胞活跃的合成功能。
2.HLA-II类抗原的表达正常皮肤及正常瘢痕的成纤维细胞并不表达HLA-II类抗原。但在硬皮病、瘢痕疙瘩和肥厚性瘢痕中,可见许多表达HLA-II类抗原的成纤维细胞。由于HLA-II类抗原为递呈特定抗原触发免疫应答所必需,推测在上述疾病中,表达HLA-II类抗原的成纤维细胞参与了局部免疫反应。
3.细胞间粘连分子1型(ICAM-1)的表达硬皮病炎症期皮损中可见一些成纤维细胞样细胞上有ICAM-1的表达。流式细胞仪检测发现,与正常成纤维细胞相比,体外培养的硬皮病成纤维细胞中高水平表达ICAM-1的细胞比例增ICAM-1与淋巴细胞功能相关抗原1型(LFA-1)是一对配体受体系统,它们可介导成纤维细胞与T细胞结合。高水平表达ICAM-1的成纤维细胞可促使拥有LFA-1的MHC-II类限制性T辅助细胞的活化。推测由此产生能促进成纤维细胞合成细胞外基质成分的细胞因子。
(二)肌纤维母细胞
在肥厚性瘢痕以及其它一些病理情况下,存着一种具有成纤维细胞和平滑肌细胞超微结构特征的细胞,即肌纤维母细胞。其起源尚不明确。利用不同细胞骨架蛋白(波形纤维蛋白、结蛋白、α-平滑肌及α-横纹肌肌动蛋白、非肌性及平滑肌肌球蛋白)的抗体,对肌纤维母细胞的表型特征进行了研究,证实了异质性纤维母细胞群的存在。其一部分显示成纤维细胞的特征,而其余的则显示有不同程度的平滑肌分化的特征。
(三)单一核浸润细胞
在硬皮病、瘢痕疙瘩及肥厚性瘢痕中,常有不同程度单一核浸润细胞增加,弥散分布或位于血管周围。应用淋巴细胞和单核细胞的单克隆抗体检测发现,主要为T淋巴细胞和巨噬细胞。这些细胞可能通过释放某些与纤维有关的细胞因子而参与真皮纤维化的发生。此外还发现,硬皮病皮损中血管周围浸润细胞处有ICAM-1和整合素(integin)β1和β2(细胞外基质在细胞表面的受体)的免疫染色强度增加。这些细胞粘连分子在淋巴细胞迁移、定位以及粘附在细胞外基质上起重要作用。
(四)内皮细胞和肥大细胞
硬皮病皮损中,内皮细胞上ICAM-1和内皮细胞白细胞连分子1型(ELAM-1)免疫染色阳性。这两种分子的表达是内皮细胞活化的表现,它们可能与淋巴细胞附着在活化的内皮细胞上有关。硬皮病、瘢痕疙瘩和肥厚性瘢痕中,肥大细胞与纤维化也有密切关系。肥大细胞(MC)根据它所含中性蛋白酶的组成和含量可分为两型,一种含类胰蛋白酶(tryptase),而胃促胰酶(chymase)含量极少,即MCT;另一种则含这两种酶,即MCTC。应用这两种酶的单克隆抗体,对硬皮病皮损区MC的数量、表型和分布进行研究,结果提示MC参与了该病的发生。
三、检测与纤维发生有关的细胞因子
转化生长因子β(TGF-β)和血小板衍生的生长因子(PDGF)与纤维化发生密切相关。用免疫细胞化学技术,在硬皮病皮损中检出了PDGF及其β型受体和TGF-β的存在,在瘢痕疙瘩皮损中也检出了TGF-β的存在。
四、其它
免疫荧光法是检测自身抗体的常用方法。系统性硬皮病患者血清中常存在多种自身抗体。在瘢痕疙瘩患者中也发现了抗成纤维细胞的抗核抗体。免疫荧光法还显示,瘢痕疙瘩和肥厚性瘢痕组织中IgM、IgG和IgA水平增高。瘢痕疙瘩与免疫的关系已引起注意并有待进一步研究。
第七节 真皮浸润细胞免疫表达
一、T淋巴细胞及其亚群的免疫表达
CD3(OKT3,Leu4)对T细胞特异,正常或肿瘤性B细胞不表达CD3;CD4RO(UCHL1)大多数T细胞和T细胞淋巴瘤呈阳性表达,能用于石蜡切片中。二者是常规筛选T细胞的良好标记物。成熟T细胞可分为表达CD4(OKT4、Leu3a)T辅助(TH/I)细胞、CD8(OKT8,Leu2a)T抑制(TS/C)细胞二种亚群。
在炎症性皮肤病中,红斑狼疮可能与T淋巴细胞介导的细胞免疫有关,红斑狼疮皮损浸润细胞大多为T细胞,最初报告TH、TS比率接近,有些报导浸润细胞多为TH;也有TS稍占优势者。大多数结果显示TS细胞较正常增加,TH和TS数较接近。因此TS细胞可能与皮损的发病机制方面有关。在扁平苔藓中浸润细胞主要是T淋巴细胞,早期TH细胞增加,而晚期,病程较长或未经治疗者TS细胞升高,TH/TS比率下降。在银屑病、尖镜湿疣、异位性皮炎皮损中,真皮浸润细胞多为T淋巴细胞,以TH为主,TS仅占少数,TH/TS比率显著增高。而银屑病、尖锐湿疣TS为主。
皮肤T细胞淋巴瘤(CTCL)具有特征性临床表现和病理变化,免疫标记显示肿瘤细胞特异性表达CD3,同时可表达CD4和CD8二种T细胞亚群标记。通常情况下肿瘤细胞以TH细胞表型为主,TS仅占少数,如蕈样肉芽肿,Sezary综合症等。由于病情的变化还可表现为TH和TS比率的改变呈TS表型为主;某种T细胞标记的缺失,如TS阴性;TH和TS标记在不成熟的T细胞中共存,呈双标记;出现不成熟的细胞标记和一种新的标记,如CD38;或不成熟细胞标记的肿瘤细胞数量增多。在诊断中T细胞标记物不能特异性区分正常和肿瘤性T细胞,在T或非T细胞性肿瘤中常出现许多反应性T细胞,而T细胞性肿瘤的组织形态可表现为多形性,在诊断阳性细胞是肿瘤性成为反应性T细胞会发生困难。由于T细胞性淋巴瘤有异常免疫表型,常缺失或不表达某一种T细胞亚群抗原,可用来鉴别肿瘤性与反应性T细胞亚群,但有时也不可能完全达到理想的诊断目的。
二、B淋巴细胞和免疫球蛋白的表达
CD19(B4)、CD20(B1)、CD22(Leu14):三种抗体能检测前B细胞和B细胞的表面抗原,但不能检测浆细胞;L26:与大多数B淋巴细胞反应,特异性强并能用于石蜡切片。B细胞在分化的大多数阶段表面有免疫球蛋白(SIg)分子,每个正常B细胞在分化的不同阶段或同一阶段上可产生几种不同类型的重链IgM(μ)、IgD(δ)、IgG(γ)、IgA(α),B细胞成熟为浆细胞则失去SIg出现胞浆内免疫球蛋白(CIg),但轻链都为同一型,κ或λ。SIg、CIg和κ、λ可作为进一步免疫表型分析。在正常B细胞群体中轻链κ和λ比例为2:1。在皮肤B细胞淋巴瘤(CBCL)中,不同的肿瘤细胞产生的SIg不同,每种B细胞可产生一种或两种重链,但只产生一种轻链,呈单克隆性。如在SIg中可见IgM和/或IgG,以其中一种阳性为主,但只有一种轻链κ或λ阳性出现,或占绝对优势。在淋巴组织反应性增生时各种B淋巴细胞都增生,呈多克隆性,可产生两种轻链和一组重链。因此,Ig能证实B细胞来源,并根据不同表达可作进一步分型。而用轻链的单一型标记可鉴别CBCL和淋巴组织反应性增生。
三、其它
CD11b(OKM1)、CD14(OKM2)、CD15(LeuM1):存在于单核细胞内;MAC387:能用于石蜡切片,单核巨噬细胞系统中许多细胞,如反应性组织细胞,上皮样细胞和巨细胞;肿瘤中浸润的巨噬细胞等均能表达MAC387,如组织细胞性淋巴瘤中瘤细胞阳性。溶菌酶:存在于单核-巨噬细胞和粒细胞中的溶酶体酶。不存在于B或T细胞。可作为真性组织细胞性淋巴瘤的标记物,但需注意在以上标记物中,肿瘤中反应增生的组织细胞可呈阳性反应,因此在判别阳性细胞是肿瘤性组织细胞还是反应性细胞,需结合形态和其它标记物予以鉴别。
第八节 汗腺及汗腺肿瘤的免疫细胞化学特点
目前已有多种PcAb和McAb用于汗腺及汗腺肿瘤的研究,这些不同的抗体对汗腺的不同部位有特异性反应,主要用于探讨汗腺肿瘤的组织发生,分化特点以及诊断及鉴别诊断。同时也可用于外科手术切除时,迅速判断肿瘤是否切除干净。现将不同的抗体分述如下。
1.癌胚抗原(CEA) 出已证明在大小汗腺的腺体及导管部均有CEA。在各种汗腺的良恶性肿瘤中也有不同的表达形式。CEA在汗腺肿瘤导管样腔隙内及其附近染色最强,汗管瘤及多形性汗腺瘤通常含有丰富的CEA,而实体性肿瘤,例如小汗腺孔瘤、螺旋腺瘤及圆柱瘤中则仅见稀疏灶状沉积,多种汗腺起源的恶性肿瘤中也见灶状CEA沉积,但CEA不能区别是起源于大汗腺或小汗腺。CEA染色不仅可研究肿瘤的发生,还可用于显微镜下的外科手术,用以快速判断肿瘤组织是否切除干净。因Paget细胞也含有CEA,因此也是鉴别Paget氏病的Paget细胞与皮肤原位癌中Paget样细胞以及恶性黑色素瘤中的Paget样瘤细胞的一种简单易行的方法。此外,CEA也存在于胎儿的小汗腺中,因此,可作为汗腺分化的最早标志。
2.S-100蛋白在皮肤病中,可用以检测Schwann氏细胞、皮肤神经黑色素细胞、痣细胞以及来源于这些细胞或组织的肿瘤。因此可用以区别梭形细胞恶性黑色素瘤与梭形细胞鳞状细胞癌以及恶性纤维组织胞瘤,用以鉴别神经来源的肿瘤与其他纤维性肿瘤,近来也有报告用S-100蛋白染早期麻风皮损中的神经组织,以了解神经的破坏,对麻风的早期诊断有一定的帮助。在小汗腺腺细胞及郎格罕氏细胞,S-100也为阳性,但小汗腺导管及大汗腺则无S-100。因此可以通过S-100染色了解汗腺肿瘤的起源,同时也有助于明确诊断。最近报告汗囊瘤中部分细胞S-100阳性,也说明这些肿瘤的组织发生与小汗腺中含有S-100蛋白的细胞有关。
3.角蛋白在大小汗腺的导管、腺体以及汗腺肿瘤中均有角蛋白存在。但对不同分子量的角蛋白则有不同的染色形式。近来报告,用识别分子量56kD的Dako-CKl及识别分子量39、43用50kD的Cam5.2二种抗体检测多种汗腺肿瘤,包括汗孔瘤、透明细胞汗腺瘤、螺旋腺瘤、皮肤混合瘤、圆柱瘤、生乳头汗管囊腺瘤及大小汗腺的汗囊瘤等,大多数肿瘤对这二种抗体呈阳性反应。也有用抗角蛋白抗体检测了多种小汗腺瘤的报告,结果所有的瘤均呈形式不同的呈阳性结果。但有些作者的结果表明汗管孔瘤、部分小汗腺孔瘤、螺旋腺瘤及大汗腺汗囊瘤等对抗角蛋白抗体EKH5呈阴性。
4.上皮膜抗原(EMA)文献报告大小汗腺的腺体部分在EMA抗原,而导管则无该抗原。某些汗腺来源的肿瘤对EMA也呈阳性反应,有报告90%的Paget氏病及乳腺导管癌EMA染色阳性。有人用EMA检测了多种小汗腺癌,包括小汗腺导管癌、透明细胞汗腺癌、乳头状汗腺癌、汗孔癌、腺样囊性癌、微囊性附件癌及小汗腺粘液腺癌等,结果均为阳性。
5.β2微球蛋白(β2MG)β2MG在多种良恶性汗腺肿瘤中可有程度不同的阳性反应。文献报告大汗腺囊瘤、乳头汗腺瘤、透明细胞汗腺瘤、真皮内导管瘤、汗管瘤、螺旋腺瘤及圆柱瘤均为阳性。部分小汗腺癌也含有这种蛋白。
6.巨囊病液体蛋白(GCDEP)GCDEP主要存在于大汗腺及大汗腺来源的多种肿瘤中,例如大汗腺汗囊瘤、乳头状汗腺瘤、生乳头汗管囊腺瘤、部分圆柱瘤及皮肤混合瘤中,也用于乳房及乳房外Paget氏病的检测。因此,GCDFP可以用于区别大小汗腺分化,有助于了解汗腺肿瘤的组织发生以及诊断及鉴别诊断。
7.表皮生长因子(EGF)EGF存在于汗腺的腺体及导管部。在某些汗腺肿瘤中也有不同的分布形式。可见于透明细胞汗腺瘤的管状上皮细胞、大汗腺混合瘤的管状及导管细胞的顶端部。
8.Peptidylarginine deiminase(PD)PD见天大小汗腺的腺体及肌上皮细胞浆中,也存在于乳房外Paget细胞内。认为该酶可作为汗腺肿瘤分类的新的标志。
9.碳酸酐酶(CA)存在于大小汗腺的腺体细胞及导管上皮细胞。可见于皮肤混合瘤的管状、导管样及囊状结构的管腔细胞。
10.锌α2糖蛋白(An2GP)An2GP主要存在于大汗腺体及导管部,部分见于小汗腺腺体,小汗腺导管则为阴性。在某些大汗腺肿瘤中多为阳性,例如大汗腺汗囊瘤及生乳头汗和囊腺瘤等。部分螺旋腺瘤及透明细胞汗腺瘤也含有这种蛋白。Zn2GP染色对于了解某些汗腺肿瘤的组织发生及分类有一定的帮助。
11.乳液脂肪球蛋白(MFG)MFG位于大小汗腺的腺体,而导管则无此蛋白,有报告90%的乳腺导管及乳房Paget氏病中MFG染色呈阳性反应,认为MFG染色有助于了解某些肿瘤的组织发生。
12.转铁蛋白(Ft )小汗腺导管外层细胞存在有FT,而末端汗管及小汗腺腺体则无此蛋白。在某些汗腺肿瘤中FT也有不同的分布形式,可见于汗管瘤细胞条索的外层细胞,在末端汗管瘤及某些汗腺癌中可呈弥漫性分布。
总之,可用于汗腺及汗腺肿瘤标记的分子很多,除上述之外,其他例如抗唾液腺酶、抗α—胰酶蛋白、抗大汗腺上皮抗原(AEA)等,对于了解汗腺肿瘤的起源及分类均有一定的帮助,但目前尚没有一种标记能区别良恶性肿瘤,或区别原发及转移性肿瘤,因此对于汗腺肿瘤的诊断,分类及组织发生,还要结合常规组织病理改变及临床特点等多方面因素进行综合考虑。
皮脂腺的免疫细胞化学特点。
目前可用于标记皮脂腺及皮脂腺肿瘤的抗体或酶有多种,包括角蛋白、EMA、MFG、CA及AEA等,但没有一种标记是皮脂腺所特有的。β2MG在皮脂腺痣、皮脂腺腺瘤及皮脂腺癌中也可呈阳性反应。近来报告皮脂腺癌中也存在OKMS抗原、虽然其性质仍不清楚,但OKMS染色有助于该病的诊断。
(孙建方 曾学思 刘季和)
参考文献
1.郭定九易凡,译.皮肤免疫病理学.1980年,长沙
2.SerreG, Viocent C , et al. Natural IgM and Igg autoantibodies to epidermal keratinsin normal human sera:I ELISA titration immunofluorecence study. J Invest Dermatol, 1987, 88:21~27
3.王刚,等.抗角蛋白自身抗体的研究进展.国外医学(皮肤病学分册),1982,18(2):74
4.聂祝湘,等.天疱疮抗体及其临床意义.临床皮肤病科杂志,1991,20(6):317~319
5. 聂祝湘,等.天疱疮治疗的某些进展.国外医学(皮肤病学分册),1991, 17(5):265~268
6.ThompsonC, et al. Expression of blood group antigen by cultured human epidermal cells.J Invest Dermatol, 1986, 86:394~398
7.jANdBos. sis . keratinocyte p 75, CTC Press, Inc . Florida AM, 1989
8. 白兆霞,等.尖锐湿疣病损的免疫组化研究.中华皮肤科杂志,1992, 25:247
9.赵玉铭,等.HLA抗原在正常皮肤中的分布.中华皮肤科杂志,1991, 23(6):402
10. VanLis JM , et al. Cin- Areaction with plasma membrane in human epidermsi. Am JClin Pathol, 1983, 779(1):143~4
11.Hashimoto K, etal. A Lectin – binding glyciprotein of Mr 135000 associated withbasal keratinocyte in pig epidermis. Biochem J, 1986,237(2):405~414
12.唐书谦,等.不同底物对大疱性皮肤病间接免疫荧光的评价评价.中华皮肤科杂志,1988,22(1):36
13. 刘荣卿,等.天疱疮及大疱性类天疱疮外观正常皮肤的免疫荧光检查.中华皮肤科杂志,1987,20:138
14. 聂祝湘,等.止血芳酸治疗天疱疮的实验观察.中华皮肤科杂志,1992, 25(4):256~258
15. WickMR and Scheithauer. Primary neuroendocrine carcinoma of the skin. In :Pathology oof Unsual MalignantCutaneous Tumors. Wick MR ed. Ne W Yorh. Marcel Dekker INC, 1985. 107
16.Narisawa Y, Hashimoto K, Bayless T, et al. Cytokeratin polypeptide Profile ofmerkel cells in human fetal and adult skin:difference of expression of cytokeratins inepidermal and dermal merkle cells. j Invest Dermatol, 1992, 98:171
17.Narisawa Y , Hashimoto K, Nihei Y, et al. Biological significance of dermalmerkle cells in development of cutaneous nerves in human fetal skin. J HistchemCytochem, 1992;40:65
18. 刘荣卿唐书谦徐洁芳,等.天疱疮及大疱性类天疱疮外观正常皮肤的免疫荧光检查.中华皮肤科杂志,1987,20(3):138~140
19. 刁庆春刘荣卿唐书谦.天疱疮、类天疱疮自身抗体检测的临床意义。中华皮肤科杂志,1988,21(5):283
20.朱学骏niimi YJ Bystryn JC.大疱性类天疱疮抗原分子异质性。中华皮肤科杂志,1990,23:291~293
21.靳培英是元甫。疱疹样皮炎在我国的发病情况及其诊断标准的商榷.中华皮肤科杂志,1991,24(4):232~235
22.Prost G, et al. Diagnosis of adultlinear IgA dermatosis by immuno ╟ electromicroscopy in 16 patients withlinear IgA deposits. J Invest Dermatol,1989, 92(1):39~45
23.wojnarowska F, Marsden RA, Bhogal B, et al. Chronic bullous disease ofchildhood, childhood cicatricial pemphigoif, and linear Iga disease of adults.A comparative study demonsrating clinical and immunopathologic overlap. J AmAcad Dermatol, 1988, 19(5):792~805
24. 朱学骏Niimi YJ Bystryn JC.抗基底膜带抗体阳性血清患者中获得性大疱性表皮松解症的发生率.临床皮肤科杂志,1990,19(4):170~173
25.Akutsu Y and Jimbow K. Immunoelectron microscopic demonstration of humanmelanosome associated antigens (HMSA) on melanoma cells:Comparison with tyrosinasedistribution. J Invest Dermatol, 1988, 90:179
26.NakanishiT and Hashimoto K. The differential reactive of benign and malignantnevomelanecytic lesions with mouse monoclonal antibody TNKH1. Cancer, 1987, 50:1340
27. ImamA, Mitchell MS, Modlin RL, et al. Human monoclonal antibodies that distinguishcutaneous malignant melanomas form benign nevi in fixed Tissue sections. JInvest Dermatol, 1986, 86:145
28. GownAM, Vogel AM, Hoak M , et al. Monoclonan antibodies specific for melanocytictumors distinguish subpopulations of melanocytes. Am j Pathol, 1986, 123:195
29.Schaumburg - Lever G, Metzler G, Kaiserling E.Ultrastructural localization ofHMB –45 binding sites. J Cutan Pathol, 1991,18:432
30. 天高文叶庆佾刘荣卿.异常黑素小体在恶性黑素瘤诊断中的意义.第三军医大学学报,1992,14(4):323
31.Furukawa K and Lloyd KO. Gangliosides in melanoma. IN:Human Melanoma. From Basicresearch to dinical application. Ferrone S ed. New York Springer –Verlag, 1990:15
32.Taniguchi M, Kuwabara I, Harada Y, et al. Cross ╟ species melanoma antigen. 1990.
33.Werkmeister JA, Triglia T, Mackay IR, et al. Fluctuations in the expression ofa glyolipid antigen assocted with differentiation of melanoma cells monitoredby a monoclonal antibody , Leo Me13. Cancer Res, 1987,47:225
34. 田芳金伯泉黄传书,等.抗人黑素瘤单克隆抗体免疫组化的研究.单克隆抗体通迅,1991,7:21
35. EstinCD, Plowman CD, Rose TM, et al. Characterization of P97, a humanmelanoma -associated antigen, and development of a recombinant vaccien formelanoma. In:Human Melanoma. From basic research to clinical application . FerroneSed. New York. Springer –Verlag, 1990.87
36.高天文刘荣卿叶庆佾.无色素性恶性黑素瘤免疫组化诊断研究.中华皮肤科杂志,1992, 25:222
37. 刘荣卿高天文.无色素性恶性黑素瘤的诊断探讨.实用肿瘤杂志,1992,7:152
38.NagataH, Ueki H, Moriguchi T, et al. Fibronectin,localization in normal human skin,granulation tissue, hypertropic scar, maure scar, progressive systemicsclerotic skin, and other fibrosing dermatoses. Arch Dermatol, 1985, 121(8):995~999
39.Linares HA. Protroglycan – lymphocyte association in the development ofhypertrophic scars. Burns, 1990, 16(1):21~24
40.Branchet MC, Boisnic S, Bletry O, et al. Exxpression of HLA class II antigensonskin fibroblasts in scleroderma. Br J Dermatol, 1992,12(5):431~435
41.CastagnoliC, Stella M, Magliacani G, et al. Anomalous expression of HLA class IImolecules on kerattinocytes and fibroblasts in hypdrtropgicscars consequent tothermal injury. Clin Exp Immunol, 1990, 82(2):350~354
42.Needleman BW, Increased expression of intercellular adhesion molecule 1 on thefibroblasts of scleroderma patientl. Arthritis Rheum,1990, 33(12):1847~1851
43.Skalli U, Schurch W, Seemayer T, et al. Myofibrollasts from perse pathologicsetting are heterogeneous in their content of actin isoforms and intermediatefilament proteins. Lab Invest, 1989, 60(2):275
44.Sollberg S, Peltonen J, Uitto J, et al. Elevated expressoin of β2and β2integrins, intercellularadhesion molecule 1, and endothelial leukocyte adhesion molecule 1 in the skinof patients with sysytemic sclerosis of recent onset .Arthritis Rheum, 1992;35(3):290~298
45. GayS, Jones RE, Huang GQ, et al. Immunohistologic demonstration of platelet -derives growth factor and sis – oncogene expression in scleroderma. J InvestDermatol, 1989, 92(2):301~303
46.Klareskog L, Gustafsson R, Scheynius A, et al. Increased expression of platelet– derives growth factor type B receptors in the skin of patients with systemicsclerosis. Arthritis Rheum, 1990, 94(2):197~203
47.Gruschwitz M, Muller PU, Sepp N, et al. Transcription and expression oftransforming growth factor type beta in the skin of progressive systemicsclerosis:amediator of fibrosis? J Invest Dermatol, 1990, 94(2):197~203
48.Peltonen J, Hsiao LL, Jaakkola S, et al. Activation of collagengene expressionin keloids :colocalization of type I and IV collagen and transforming growth factor–β1mRNA.J Invest Dermatol, 1991;97(2):240~248
49.TraniAMA, Gruber BL, Kaufman LD, et al. Mast cell changes in scleroderma. ArthritisRheum, 1992;35(8):933~939
50.Hashimoto K, et al. Anti – keratin monoclonal antibodies :production, specificities andapplications. J Cutan Pathol, 1983;6(10):529~539
51.KallioinenM, et al. β-2-microglobulin in benign and malignant adnexal skin tumors andmetastasizing basocellular carcinoma. J Cutan Pathol, 1984;1(11):27~34
52.Penney NS. Immunohistochemistry of adnexal neoplasms. j Cutan Pathol , 1984;(11):357~364
53.SwansonPE, et al. Eccrine sweat gland carcarcinoma:an histologic and immunohistochemical studyof 32 cases. J Cutan Pathol,1987,2(4):65~86
54. NodaY, et al. Immunohistochemical study of carbonic anhydrase in mixed tumors andadenomas of sweat and sebaceous glands. J Cutan Pathol, 1987;5(14):285~290
55.NodaY,et al. Immunohistochemical localization by monoclonal antibody of humanepidermal growth factor in mixed tumors of the skin. J Cutan Pathol, 1987;6(14):337~342
56. ZukJA, et al. Immunohistochemistry staining patterns of sweat glands and theirneoplasms using two nomoclonal antibodies to keratieos. j Cutan Pathol, 1988;1(15):8~17
57.MazoujianG, et al. Immunohistochemistry of gross disease fluid protein (GCDFP-15) in 65benign sweat gland tumors of the skin. Am j Dermatopathol, 1988, 1(10):28~35
58. WoodWS, et al. Mammary Paget’s disease and intraductal carcinoma :histologic, histologic,histochemical and immunocytochemical comparison. Am j Dermatopathol, 1988,10(3):188
59.Mihara M. Chondroid syringoma associated with hidrocystoma – like changes :possible differentiation intoeccring gland:a histologic. immunohistochemical and electron microscopic study. JCutan Pathol, 1989, 5(16):281~286
60.Penneys NS, et al. Immunohistochemistry demonstration of ferritin in sweatgland and sweat gland neoplasms. J Cutan Pathol, 1990, 1(17):32~36
61. UranoY, et al. Immunohistochemistry demonstration of petidylarginine deiminase inhuman sweat glands. Am J Dermatopathol, 1990;12(3):249~255
62.Mazoujian G. Immunohistochemistry of GCDFP –24 and zinic alpha2 glycoprotein inbenign sweat gland tumors. Am J Dermatopathol,1990, 12(5):452~457
第十八章 核酸分子杂交技术概述
第一节 核酸的分子结构
一、核酸的化学组成
组成核酸的元素有C、H、O、N、P等,其中N含量约为15%~16%,磷含量为9%~10%。由于核酸分子中的磷含量比较恒定,因此,核酸定量测定的经典方法,是以测定磷含量代表核酸量。
核酸经水解可得到多核苷核,因此核苷酸是核酸的基本单位,核酸就是由很多单核苷酸聚合形成的多核苷酸,核苷酸可被水解产生核苷和磷酸,核苷还可进一步水解,产生戊糖和含氮碱。因此,核酸是由含氮碱、戊糖及磷酸三种成分组成。
含氮碱(简称碱基):核酸中的含氮碱简称碱基,是嘌呤碱(purine)与嘧啶碱(pyrimidine)的衍生物。RNA和DNA含有的共同碱基成分是腺嘌呤(adenine,A)、鸟嘌呤(guanine,G)和胞嘧啶(cytosine, C)。二者的区别是RNA含有尿嘧啶(uracil,U),而DNA含有胸腺嘧啶(thymine,T)。嘌呤和嘧啶都有酮-烯醇式互变异构现象,一般生理pH条件下呈酮式。它们的结构如下:
有些核酸中含有修饰碱基(或稀有碱基),这些碱基大多是在上述嘌呤或嘧啶碱的不同部位甲基化(methylation)或进行其它的化学修饰而形成的衍生物。例如有些DNA分子中含有5-甲基胞嘧啶(m5C),5-羟甲基胞嘧啶(hm5C)。某些RNA分子中含有1-甲基腺嘌呤(m1A)、2,2-二甲基鸟嘌呤(m22G)和5,6-二氢尿嘧啶(DHU)等。
嘌呤碱和嘧啶碱一般多不易溶于水,对250~280nm波长的紫外光有较强的吸收,但对260nm光波的吸收能力最大。由于碱基是核酸的基本组成成分,因此,所有的核酸(包括DNA和RNA)其共同特点是对260nm处的紫外光有最大的吸收值。
核酸分子中碱基的克分子数与磷的克原子数相等,所以可根据核酸溶液中的磷含量及紫外光的吸收值来测定核酸量。一般以每升核酸溶液中含1g磷原子为标准来计算核酸的吸光率,这称为克原子磷吸光率或克原子磷消光系数[ε(p)],ε(p)的计算式为:
ε(p)=A/Cl
A为吸光度(光密度),1为比色杯内径,通常为1.0cm;C为每升核酸溶液中磷的克原子数。
C= 每升溶液中磷重(wg)/30.98ε(p)=30.98A/wl
一般DNA的ε(p)=6000~8000;RNA为7000~10000
现将DNA和RNA的化学组成归纳如表18-1:
表18-1
| RNA | DNA | |
| 嘌呤碱 | 腺嘌呤鸟嘌呤 | 腺嘌呤鸟嘌呤 |
| 嘧啶碱 | 胞嘧啶尿嘧啶 | 胞嘧啶胸腺嘧啶(或5-甲基胞嘧啶) |
| 戊糖 | 核糖 | 脱氧核糖 |
| 磷酸 | 磷酸 | 磷酸 |
二、核酸的一级结构
核酸又称多核苷酸,组成DNA的脱氧核糖核苷酸主要为四种,即dAMP、dGMP、dCMP及dTMP;组成RNA的核糖核苷酸主要有AMP、GMP、CMP及UMP四种。对两种核酸的组成可简写如下:
DNA=(碱基-脱氧核糖-磷酸)n;RNA=(碱基-核糖-磷酸)n
核酸中核苷酸的连接方式为:一个核苷核C-3’上羟基与下一个核苷核酸C-5’连接在磷酸羟基脱水缩合成酯键,称酯键称3’5’磷酸二酯键,若干个核苷酸间依3’5’磷酸二酯键连接成长链的大分子即为核酸。此长链称多核苷酸链,在链的一核苷酸,其C-5’连接的磷酸只一个酯键,称此核苷酸为链的5’磷酸未端或5’。链的另一端核苷酸上C-3’上羟基是自由的,对此核苷酸称为3’羟基末端或3’端,链内的核苷酸在C-5’上磷酸已形成二酯键,C-3’上羟基也已参与二酯键的形成,故称核苷酸残基。
核酸的一级结构乃指其核苷酸链中核苷酸的排列顺序,由于核酸中核苷酸彼此之间的差别乃在于碱基部分,故核酸的一级结构即指核酸分子中碱基的排列顺序。
对核酸一级结构的描述为:将5’磷酸末端书于左侧,中间部分为核苷酸残基,3’羟基末端书于右侧。通常用竖线表示核糖,碱基标于竖线上端,竖线间有含P的斜线,代表3’,5’磷酸二酯键。此表示法及简化式如下:
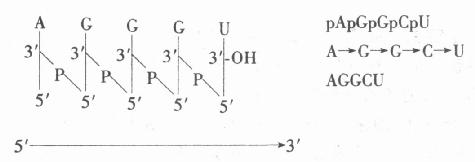
三、核酸的高级结构
核酸的多核苷酸链在次级键的基础上,还可形成更为复杂的二级及三级的高级结构。
(一)DNA
1.二级结构1953年Watson 及Crick在化学分析及X光衍射法观察DNA结构的基础上提出了著名的DNA双螺旋结构模型(double helix model)此结构是在核酸一级结构基础上形成的更为复杂的高级结构,即DNA的二级结构,结构如图18-1。
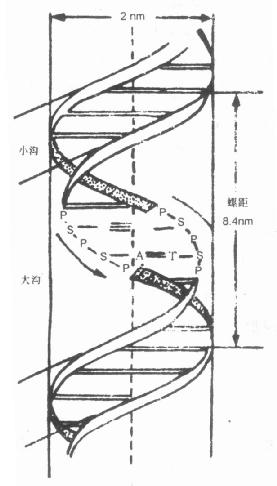
图18-1 DNA的双螺旋结构
P:磷酸基;S:脱氧核糖;G:鸟嘌呤;
A:腺嘌呤;T:胸腺嘧啶C:胞嘧啶
DNA的二级结构即双螺旋结构,其内容可归纳为:
(1)DNA分子为二条多核苷酸链以一共同轴为中心,盘绕成右手双螺旋结构。螺旋直径2nm。螺旋盘绕形成链间的两种沟,即宽的大沟与狭窄的小沟。
(2)二条多核苷酸链的走向相反,通常取左侧链从上到下为5’→3’端,右则链从下向上为5’→3’端,这样二条链构成反平行排列的双螺旋。
(3)二条多核苷酸链借氢键而连系在一起。氢键乃一链碱基上-NH2的氢与另一链上碱基的氧或氮形成。碱基有二个氢键,G与C之间有三个氢键(图18-3)。这种相配关系称为碱基互补或碱基配对。配对的碱基处于同一平面,此平面与双螺旋的中心轴垂直,由于二条链中碱基互补,所以二链彼此又称为互补链。
(4)碱基对之间氢键的能量为3~7kcal/mol,由于氢键多,所以可维系DNA双链结构。另外碱基对彼此间距离为0.34nm,每一螺旋含10个碱基对,故螺距为3.4nm,相邻碱基对间彼此尚有范德瓦士(van der Waals)力作用(此力量为1~2kcal/mol,作用范围为0.5nm),能量虽弱但由于碱基对多,合力也就大。可见碱基对的氢键及碱基对之间的范德瓦士力是稳定DNA成双螺旋结构的主要能量。
上述DNA的双螺旋结构是溶液及活体中常见的形式,通称B型。当B型所处条件的湿度低于75%时,可转变为A型。A型的碱基对不垂直于双螺旋的轴、倾斜约20度,螺距降为2.8nm,每一螺旋含11个碱基对。B型与A型的水合程度不同,它们是DNA分子在天然条件下的两种基本形式。除A、B型外尚发出有C型双螺旋,似B型,螺距3.3nm,第一螺旋含9个碱基对。
DNA双螺旋结构阐明的量重要意义在于第一次提出了遗传信息是以DNA分子中核苷酸的排列顺序为储存方式,从而说明了天然遗传信息的复制过程。
2.三级结构已发现线粒体、叶绿体、细菌、质粒及一些病毒的DNA双螺旋分子尚可形成封闭环状,天然状态的环状DNA分子多扭曲成麻花状的超螺旋结构(superhelix),这些比螺旋更为复杂的结构即DNA分子的三级结构。
真核生理细胞核中的DNA具有一种超螺旋结构,即DNA双螺旋盘绕在组蛋白上形成核小体(nucleosome)。核小体是染色质(chromatin)的核心小粒,由有140个碱基对的双螺旋DNA缠绕于由组蛋白(H2A、H2B、H3及H4各二分子)组成的八聚体外面,这一DNA股由此形成直径为9nm的超螺旋1.75圈。此核小体又经60个碱基对的DNA双螺旋及组蛋白H1形成细丝(间隔区)与下一个核小体相连接。
核小体的DNA双螺旋为200个碱基对,长度应为0.34nm×200=68nm,但实际长度只10nm,说明DNA双螺旋链进一步螺旋化盘绕在组蛋白八聚体上,其长度压缩了7/8。每6个核小体又绕成一圈形成螺线管,外径为30nm,螺距为10nm。这样DNA分子长度被压缩了6/7。120个螺线管又盘绕成直径为400nm,高为30nm的超螺线管,DNA分子长度又被压缩了40/41,此超螺线管即染色体的单位纤维(unit fiber),长20~60nm。从单位纤维形成染色单体(chromatid),实际长度为2~10nm,DNA分子长度又被压缩了5/6~6/7。这样从许多核小体组成的串珠样纤维经多层次螺旋化结构到形成染色单体,DNA分子的长度已被压缩至近1/10000。
(二)RNA
RNA分子也是由核苷酸依3’,5’磷酸二酯键形成的多核苷酸链。RNA总是以单链的形式存在,也有5’磷末端及3’羟基末端。RNA单链的局部折叠成的某一片段的A及G分别与另一片段的U及C配对常形成发夹结构(hairpin structure)。在此结构内的碱基无需全部配对,而配对部位形成小的双螺旋区域,不能配对的碱基则连成小环从螺旋区中被圈出来。这种RNA单链局部小双螺旋结构即是RNA的二级结构。
四、基本组织
由于DNA分子中核苷酸序列分析以及一级结构与功能相关的研究,使得人们有可能进一步了解DNA一级结构与基因组织的关系。已经证实,自然界极大多数生物体遗传信息贮存在DNA的核苷酸排列顺序中,因此,基因是DNA的一个片段,只有少数病毒的遗传信息贮存在RNA分子中。DNA分子中不同区域有不同功能,有些区域可编码蛋白质(最终产物是蛋白质),有些区域可编码RNA(最终产物是tRNA和rRNA),有些序列则与调控有关。那么一个DNA分子能携带多少基因呢?如果以平均1000个碱基对可编码一个3kD的蛋白质计算,猴病毒(SV40)DNA分子量为3.0×105,有5000碱基对可编码5种蛋白质。人染色体DNA有2.3×109碱基对,可编码200万以上的基因,但实际上,最多可编码基因数为2~3万。这是由于真核细胞DNA分子除编码蛋白质和RNA等结构基因外,有相当部分DNA顺序属于非编码区,而原核细胞DNA由于分子较小,必需充分利用有限的核苷酸序列。各种生物体内DNA分子的大小见表18-2。
表18-2 各种生物体内DNA分子的大小
| 来源 | 分子量 | 碱基对数目 | 长度 |
| 噬菌体фX174 | 0.6μm | ||
| 腺病毒(SV40) | 1.6×106 | 4500 | 1.5μm |
| 鼠线粒体 | 3.0×106 | 14000 | 4.9μm |
| 噬菌体λ | 9.5×106 | 50000 | 17μm |
| 噬菌体T2或T4 | 3.3×107 | 200000 | 67μm |
| 大肠杆菌染色体 | 1.3×108 | 4500000 | 1.5μm |
| 人染色体 | 3.0×109 | 125000000 | 4.1μm |
1.真核生物的基因组织根据某一段核苷酸顺序在整个DNA分子中出现的频率不同可分为以下几种:
(1)单拷贝顺序(singlecopy sequence):在整个DNA分子中只出现一次或少数几次,主要是编码蛋白质的结构基因。除组蛋白、角蛋白和肌动蛋白以外,几乎所有的蛋白质基因都是单拷贝顺序,平均为1000碱基对。单拷贝基因在整个基因组织中所占比例最高。在人的细胞中约占DNA含量的一半。
(2)中等重复顺序(moderatelyrepetitive sequences):有些基因如核蛋白体RNA基因、tRNA基因、组蛋白基因等在DNA分子中可重复出现几十到几千次,约占人细胞DNA含量的30~40%。以rRNA为例,在大肠杆菌中重复频率为7而果蝇中可重复千次。可见真核细胞中重复顺序比原核细胞高得多。
(3)高重复顺序(highlyrepetitive sequences):可重复几百万次。往往是简单的重复顺序,如蟹的T-A-T-A-T-A-T。也有的较长如非洲绿猴DNA是以172个碱基对的顺序为基础重复几万次。高重复顺序一般位于异染色质上,多数不编码蛋白质或RNA,其功能还不太清楚,主要是起间隔作用,可能与调控有关。
在重复顺序中还有一种反转重复顺序(inverted repetitivesequences)。其特点是一段碱基呈现回文结构,即一条单链回折即可形成互补的双链,故称为回文结构(palindromicstructure)或发夹结构(hairpin structure)。这种结构对基因的复制与转录可能具有调节 控制功能。

真核细胞中单拷贝顺序和重复顺序常常是中间隔排列的。不仅如此,在一个基因内部往往被一个或几个额外的顺序分割成若干片段,这种插入到基因内部的顺序称为插入顺序或内含子(intron)。内含子是不编码的顺序,而编码的碱基顺序则称为外显子(exon)。插入顺序是真核细胞DNA最主要的特征。
真核生物由于存在着较多的重复顺序、特殊的插入顺序以及控制区和其它多余顺序,使得DNA总长度往往大于编码的结构基因,因此,实际基因数往往小于DNA分子。
2.原核生物的基因组织原核生物DNA分子较小,基因组织也较简单,一般具有以下特点:
(1)DNA分子绝大部分用于编码蛋白质,不编码部分(又称间隔区)通常包含控制基因表达的顺序。例如,噬菌体фX174中只有5%是非编码区。
(2)功能相关的基因常常串联在一起,并转录在同一个mRNA分子中,称为多顺序反子。这种现象在真核生物中是很少见的。
(3)基因重叠:例如фX174的E基因全部包括在D基因内,B基因则包括在查基因内。这种现象主要发现在病理DNA分子中,可能是由于DNA分子太小又要装入相当量的基因的缘故。
第二节 DNA的变性与复性
一、DNA变性
DNA变性是指双螺旋之间氢键断裂,双螺旋解开,形成单链无规则线团,因而发生性质改变(如粘度下降,沉降速度增加,浮力上升,紫外吸收增加等),称为DNA变性。加热、改变DNA溶液的pH、或受有机溶剂(如乙醇、尿素、甲酰胺及丙酰胺等)等理化因素的影响,均可使DNA变性。
通常,可利用DNA变性后波长260nm处紫外吸收的变化追踪变性过程。因为DNA在260nm处有最大吸收值这一特征是由于含有碱基组成的缘故,在DNA双螺旋结构模型中碱基藏于内侧,变性时由于双螺旋解开,于是碱基外露,260nm紫外吸收值因而增加,这一现象称为增色效应(hyperchromic effect)。见图18-2。
图18-2 DNA的增色反应
如果升高温度使DNA变性,以温度对紫外吸收作图,可得到一条曲线,称为溶解曲线(见图18-3),由图可见当温度升高到一定范围时,DNA溶液在260nm处的吸光度突然明显上升至最高值,随后即使温度继续升高,其吸光度也无明显变化。由此说明DNA变性是在一个很窄的温度范围内发生,增色效应是爆发式的。从而也说明当达到一定温度时,DNA双螺旋几乎是同时解开的。通常人们把50%DNA分子发生变性的温度称为变性温度(即熔解曲线中点对应的温度),由于这一现象和结晶的融解相类似,故又称融点或融解温度(melting temperature, Tm)。因此Tm是指消光值上升到最大消光值一半时的温度。
图18-3 DNA的Tm值
综上所述,Tm值和增色效应是目前描述DNA特性所常用的两个量。假定一个DNA大分子最初全部是双螺旋结构,在热变性后消光系数上升30%以上;如果DNA原先局部就处于单链状态(例如在分子末端),则变性后上升较少。增色效应的大小是DNA性质的一个简单指标,与分子量无关。Tm不是一个固定的数值,它与很多因素有关:pH、离子强度和DNA的碱基比例。随着溶剂内离子强度上升,Tm值也随着增大。在某一离子强度(~10-3M)以下,无需加热就使溶于其中的DNA出现不可逆变性。与A-T碱基配对比较,DNA双螺旋内的G-C配对更为牢固。在相同条件下,DNA内G-C配对含量高,其Tm值也高。
假定在一个双链DNA分子内某些片段含有较多G-C碱基对,根据它们局部Tm值差,用电子显微镜就可以观察和测量到这些片段,如在DNA某一片段内含有较多的A-T碱基对,在某一个温度时就可能出现双链解离的现象。但在同一温度下,含G-C对较多部分仍然保持双链结构。这是一种非常有用的技术。
DNA的Tm值与以下因素有关:
(1)DNA的均一性:均一DNA如病毒DNA,解链发生在很窄的范围内,而不均一的DNA如动物细胞NDA其Tm值的范围则较宽。
(2)DNA分子中(G+C)的含量:一定条件下DNA的Tm值,由G+C含量所决定,因为G+C之间有3个氢链,因此G+C含量较高的DNA,Tm值较高,二者的关系可用以下经验式表示:
%(G+C)=(Tm-63.0)×2.44
实验表明DNA分子中(G+C)克分子含量百分比的大小与Tm值的高低呈直线关系,见图18-4。
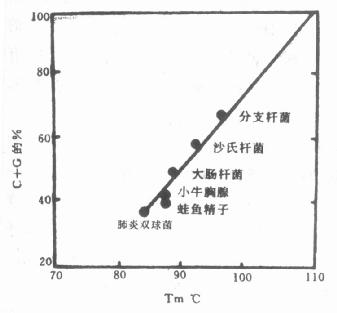
图18-4 DNA和Tm值与G-C含量的关系
(3)溶剂的性质:Tm不仅与DNA本身性质有关,而且与溶液的条件有关,通常溶液的离子强度较低时,Tm值较低,融点范围也较宽,离子强度增高时,Tm值长高,融点范围也变窄。因此,DNA制剂不应保存在离子强度过低的溶液中,一般保存在1mol/l NaCl溶液中较稳定。
二、复性
变性DNA只要消除变性条件,二条互补链还可以重新结合,恢复原来的双螺旋结构,这一过程称为复性(renaturation)。通常DNA热变性后,将温度缓慢冷却,并维持在比Tm低25~30℃左右时,变性后的单链DNA即可恢复双螺旋结构,因此,这一过程又叫做退火。复性后的DNA,理化性质都能得到恢复。倘若DNA热变后快速冷却,则不能复性(图18-5)。
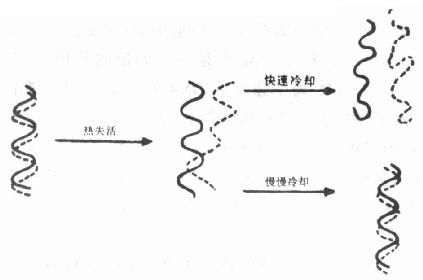
图18-5 热变性过程和两种冷却过程示意图
影响复性速度的因素很多,同样条件下,DNA顺序简单的分子复性很快,如polyd[T]和polyd[A]由于彼此互补识别很快,故能迅速复性。但顺序较复杂的DNA分子复性则较慢。因此通过变性速率的研究,可以了解DNA顺序的复杂性。DNA片段的大小也影响变性的速率,因为DNA片段愈大,扩散速度愈低,使DNA片段线状单链互相发现互补的机会减少。因此,在复性实验中,有时将DNA切成小片段,再进行复性。同样条件下,同一种DNA浓度愈高,复性速度也愈快。溶液的离子强度对复性速度也有影响,通常盐浓度较高时,复性速度较快。
Doty研究小组是最早对DNA变性过程进行深入研究的。它们所获得的结果表明,在达到Tm值时,两条DNA单链分离开。如果在加热之后慢慢地冷却,则出现部分复性,即DNA的一部分回复到双螺旋结构。复性的程度取决于DNA浓度及信息含量的多少。病毒DNA(信息含量少)比哺乳动物DNA容易复性,而DNA浓度较高时,有利于复性,快速冷却使DNA仍然处于变性状态,这时自由单链成链线团结构。对这种情况,人们称之为螺旋-线团转化过程(helix-coil-transition)。快速冷却时消光系数固然有所下降,但比天然DNA的数值始终要大。
细胞核DNA复性的动力学研究指出,DNA内很少片段有重复的或很相似的碱基顺序(所谓重复DNA)。DNA复性的程度和过程与其信息含量的多少等有关;因而病毒DNA比细菌DNA复性得快。Britten发现一种测定和观察复性过程的方法。X轴表示变性DNA原始浓度(Co)和保温时间的乘积,纵轴表示DNA复性部分(重新作为双螺旋结构出现)。DNA比例可以用羟基磷灰石柱的办法加以确定,因为这种柱能够使单链和双链DNA分离开来。DNA复性曲线呈S形,随着信息含量增加,此形状相同曲线往往较高Co.t值处移动。奇怪的是,从细胞核来的DNA在复性时显示出完全不同的情形:这些DNA中的一部分异常快地复性,而另一些DNA只有在极高的Co.t值时才出现预期的复性。对快速复性可以作这样的解释,即在某一DNA之内同时有几个相同或很类似的顺序存在,因而找重复顺序比找DNA内唯一顺序要快得多。后者含有特殊的遗传信息,常被称为独特DNA。与之相反是重复DNA片段。
真核DNA自发复性的一种特殊途径是通过发夹结构。对单链而言,要生成这种发夹结构,要求一种特定的碱基顺序,这种顺序称作回文(正读反读都相同)结构。为了构成回文结构,DNA片段的碱基顺序必须在互补链内找到相反的顺序;在具有相反碱基顺序的两个DNA片段之间,显然常常出现短的中间片段由于存在这样的核苷酸顺序,在复性时就能形成发夹结构。
如果存在很多重复回文结构,在部分复性时就能通过形成DNA侧链而出现十字结构。DNA回文结构使DNA片段出现回旋对称性。这种结构常常出现在DNA和蛋白质之间相互作用的地方,特别是后者起控制作用时。
第三节 分子杂交
一、概述
前面已经介绍了核酸分子单链之间有互补的碱基顺序,通过碱基对之间非共价键(主要是氢键)的形成即出现稳定的双链区,这是核酸分子杂交的基础。杂交分子的形成并不要求两条单链的碱基顺序完全互补,所以不同来源的核酸单链只要彼此之间有一定程度的互补顺序(即某种程度的同源性)就可以形成杂交双链。分子杂交可在DNA与DNA、RNA与RNA或RNA与DNA的二条单链之间进行。由于DNA一般都以双链形式存在,因此在进行分子杂交时,应先将双链DNA分子解聚成为单链,这一过程称为变性,一般通过加热或提高pH值来实现。使单链聚合双链的过程称为退火或复性。用分子杂交进行定性或定量分析的最有效方法是将一种核酸单链用同位素或非同位素标记成为探针,再与另一种核酸单链进行分子杂交。
核酸杂交技术基本上是Hall等1961年的工作开始的,探针与靶序列在溶液中杂交,通过平衡密度梯度离心分离杂交体。该法很慢、费力且不精确,但它开拓了核酸杂交技术的研究。Bolton等1962年设计了第一种简单的固相杂交方法,称为DNA-琼脂技术。变性DNA固定在琼脂中,DNA不能复性,但能与其它互补核酸序列杂交。典型的反应是用放射性标记的短DAN或RNA分子与胶中DNA杂交过夜,然后将胶置于柱中进行漂洗,去除游离探针,在高温、低盐条件下将结合的探针洗脱,洗脱液的放射性与结合的探针量呈正比。该法尤其适用于过量探针的饱和杂交实验。60年代末,Britten等设计了另一种分析细胞基因组的方法。该法是研究液相中DNA的复性以比较不同来源核酸的复杂度,典型的方法是:从不同生物体(细菌、酵母、鱼和哺乳动物等)内分离DNA,用水压器剪切成长约450核苷酸(nt)的片段。剪切的DNA液(含0.12mol/L磷酸盐缓冲液或0.18mol/l Na+),经煮沸使dsDNA热变性成ssDNA。然后冷至约60℃,在此温度孵育过程中,测定溶液一定时间内的UV260nm的吸光度(减色效应)来监测互补链的复性程度。通常该实验可比较不同来源生物DNA的复性速率,并可建立序列复杂度与动力学复杂度间的关系。
60年代中期Nygaard 等的研究为应用标记DNA或RNA探针检测固定在硝酸纤维素(NC)膜上的DNA序列奠定了基础。如Brown等应用这一技术评估了爪蟾rRNA基因的拷贝数。RNA在代谢过程中被3H尿嘧啶标记,并在过量的情况下与膜上固定的基因组DNA杂交,继而用RNase处理,消化非特异性结合的RNA。漂洗后计数以测定杂交探针的量。通过计算与已知量DNA杂交的RNA量即可评估rRNA基因数。由于当时缺乏特异探针,这种方法不能用于研究其它特异基因的表达,这些早期过量探针膜杂交试验实际上是现代膜杂交实验的基础。
进入70年代早期,许多重要的发展促进了核酸杂交技术的进展。例如,对特异基因转录产物的分析和对动力学杂交实验又有兴趣。固相化的Poly U –Sepharose和寡(dT)-纤维素使人们能从总RNA中分离PolyA+ RNA。用mRNA的经纯化技术可从网织红细胞总RNA中制备α-和β-珠蛋白mRNA混合物。这些珠蛋白mRNA首次被用于合成特异的探针以分析珠蛋白基因的表达。由于制备cDNA探针很繁琐,所获得cDNA的长度和纯度也不稳定。所以寻求新的探针来源是使分子杂交技术进一步推广的基础。
70年代末期到80年代早期,分子生物学技术有了突破性进展,限制性内切酶的发展和应用使分子克隆成为可能。各种载体系统的诞生,尤其是质粒和噬菌体DAN载体的构建,使特异性DNA探针的来源变得十分丰富。人们可以从基因组DNA文库和cDNA文库中获得特定基因克隆,只需培养细菌,便可提取大量的探针DNA。迄今为止,已克隆和定性了许多特异DNA探针。
由于固相化学技术和核酸自动合成仪的诞生,现在可常规制备18~100个碱基的寡核苷酸探针。应用限制酶和Southern印迹技术,用数微克DNA就可分析特异基因。特异DNA或RNA序列的量和大小均可用Southern印迹和Northern印迹来测定,与以前的技术相比,大大提高了杂交水平和可信度。
尽管取得了上述重大进展,但分子杂交技术在临床实用中仍存在不少问题,必须提高检测单拷贝基因的敏感性,用非放射性物质代替放射性同位素标记探针以及简化实验操作和缩短杂交时间,这样,就需要在以下三方面着手研究:第一,完善非放射性标记探针;第二,靶序列和探针的扩增以及信号的放大;第三,发展简单的杂交方式,只有这样,才能使DNA探针实验做到简便、快速、低廉和安全。
二、探针-靶反应
从化学和生物学意义上理解,探针是一种分子,它带有供反应后检测的合适标记物,并仅与特异靶分子反应。抗原-抗体、外源凝集素-碳水化合物、亲和素-生物素、受体-配基(ligand)以及互补核酸间的杂交均属于探针-靶分子反应。蛋白质探针(如抗体)与特异靶分子是通过混合力(疏水、离子和氢键)的作用在少数特异位点上的结合,而核酸探针与互补链的反应则是根据杂交体的长短不同,通过氢键在几十、几百甚至上千个位点上的结合。因为有机溶液可降低杂交体的稳定性,所以,疏水反应对互补核酸链的结合也有一定的作用,但对其特异性影响甚微。
核苷酸经某一原子、功能基团或长侧链修饰后仍可能进行碱基配对,这取决于修饰的部位和修饰的性质。这一特性有助于理解非放射性核酸探针标记物的设计和125I与DNA探针的化学结合。能与核酸结合的单一原子有银、溴和碘等,这些元素可与嘧啶(胸腺嘧啶除外)环的C-5位或嘌呤环的C-8位反应而不影响氢键的形成。溴亦可与胸腺嘧啶的C-6位结合。而胞嘧啶的C-4和腺嘌呤的N-6就不能被修饰,否则会影响碱基配对,尽管C的N-4位和A的N-6位参与了氢键形成,但它们也是标记位点。这是因为标记的探针每1kb只掺入10~30个修饰碱基,即仅4%~12%的单个碱基被修饰的类似物取代了。尽管掺入位点处的碱基配对较弱或不存在,但对整个杂交分子的稳定性影响很小。防止氢键破坏的一种方法就是修饰探针,即探针克隆入M13载体中,只修饰载体区而不修饰插入片段。当用放射性同位素32P和35S标记核酸时,由于同位素是掺入核酸骨架的磷酸二脂键中,因此碱基未发生任何修饰。在5’端的磷酸基团上可进行化学修饰,这是标记寡核苷酸探针的有效方法。因为这种方法是在一个探针分子上标记一个检测的基团,所以,对长的克隆探针不适用。
此外,还可利用修饰的碱基来增加杂交的稳定性和特异性。2-氢基腺嘌呤可替代寡核苷酸探针中的腺嘌呤通过形成3个氢键以增加杂交体的稳定性。另外,在G-C丰富的RNA探针中,可用次黄嘌呤代替鸟嘌呤以获得特异的杂交。因为次黄嘌呤和鸟嘌呤间只形成2个氢键,有效地降低了杂交体的Tm值,这样,Tm值与杂交温度更接近,杂交的严格性就增加了,因此,也就增加了特异性。
很显然,结合位点的不同和可检测基团与检测系统的不同,可派生出很多核酸探针标记方法。这是由核酸的化学结构和性质所决定的。只有在对核酸分子的探针-靶反应的化学本质有了深入了解之后,才能更好地理解后面的章 节 内容。
三、核酸探针的种类
基因探针根据标记方法不同可粗分为放射性探针和非放射性探针两大类,根据探针的核酸性质不同又可分为DNA探针,RNA探针,cDNA探针,cRNA探针及寡核苷酸探针等几类,DNA探针还有单链和双链之分。下面分别介绍这几种探针。
(一)DNA探针
DNA探针是最常用的核酸探针,指长度在几百碱基对以上的双链DNA或单链DNA探针。现已获得DNA探针数量很多,有细菌、病毒、原虫、真菌、动物和人类细胞DNA探针。这类探针多为某一基因的全部或部分序列,或某一非编码序列。这些DNA片段须是特异的,如细菌的毒力因子基因探针和人类Alu探针。这些DNA探针的获得有赖于分子克隆技术的发展和应用。以细菌为例,目前分子杂交技术用于细菌的分类和菌种鉴定比之G+C百分比值要准确的多,是细菌分类学的一个发展方向。加之分子杂交技术的高敏感性,分子杂交在临床微生物诊断上具有广阔的前景。细菌的基因组大小约5×106bp,约含3000个基因。各种细菌之间绝大部分DNA是相同的,要获得某细菌特异的核酸探针,通常要采取建立细菌基因组DNA文库的办法,即将细菌DNA切成小片段后分别克隆得到包含基因组的全信息的克隆库。然后用多种其它菌种的DNA作探针来筛选,产生杂交信号的克隆被剔除,最后剩下的不与任何其它细菌杂交的克隆则可能含有该细菌特异性DNA片段。将此重组质粒标记后作探针进一步鉴定,亦可经DNA序列分析鉴定其基因来源和功能。因此要得到一种特异性DNA探针,常常是比较繁琐的。探针DNA克隆的筛选也可采用血清学方法,所不同的是所建DNA文库为可表达性,克隆菌落或噬斑经裂解后释放出表达抗原,然后用来源细菌的多克隆抗血清筛选阳性克隆,所得到多个阳性克隆再经其它细菌的抗血清筛选,最后只与本细菌抗血清反应的表达克隆即含有此细菌的特异性基因片段,它所编码的蛋白是该菌种所特有的。用这种表达文库筛选得到的显然只是特定基因探针。
对于基因探针的克隆尚有更快捷的途径。这也是许多重要蛋白质的编码基因的克隆方法。该方法的第一步是分离纯化蛋白质,然后测定该蛋白的氨基或羟基末端的部分氨基酸序列,然后根据这一序列合成一套寡核苷酸探针。用此探针在DNA文库中筛选,阳性克隆即是目标蛋白的编码基因。值得一提的是真核细胞和原核细胞DNA组织有所不同。真核基因中含有非编码的内含子序列,而原核则没有。因此,真核基因组DNA探针用于检测基因表达时杂交效率要明显低于cDNA探针。
DNA探针(包括cDNA探针)的主要优点有下面三点:①这类探针多克隆在质粒载体中,可以无限繁殖,取之不尽,制备方法简便。②DNA探针不易降解(相对RNA而言),一般能有效抑制DNA酶活性。③DNA探针的标记方法较成熟,有多种方法可供选择,如缺口平移,随机引物法,PCR标记法等,能用于同位素和非同位素标记。
(二)cDNA探针
cDNA(complementary DNA)是指互补于mRNA的DNA分子。cDNA是由RNA经一种称为逆转录酶(reverse transcriptase)的DNA聚合酶催化产生的,这种逆录酶是Temin等在70年代初研究致癌RNA病毒时发现的。该酶以RNA为模板,根据碱基配对原则,按照RNA的核苷酸顺序合成DNA(其中U与A配对)。这一途径与一般遗传信息流的方向相反,故称反向转录或逆转录。携带逆转录酶的病毒侵入宿主细胞后,病毒RNA在逆转录酶的催化下转化成双链cDNA,并进而整合人宿主细胞染色体DNA分子,随宿主细胞DNA复制同时复制。这种整合的病毒基因组称为原病毒。在静止状态下,可被复制多代,但不被表达,故无毒性。一旦因某种因素刺激而被活化,则该病毒大量复制,如其带有癌基因,还可能诱发细胞癌变,后来发现逆转录酶不仅普遍存在于RNA病毒中,而且哺乳动物的胚胎细胞和正在分裂的淋巴细胞也含有逆转录酶。逆转录酶的作用是以dNTP为底物,RNA为模板,tRNA(主要是色氨酸tRNA)为引物,在Trna3’-OH末端上,5’-3’方向,合成与RNA互补的DNA单链,称为互补DNA(cDNA),单链cDNA与模板RNA形成RNA-DNA杂交体。随后在逆转录酶的RNase H活性作用下,将RNA链水解成小片段。cDNA单链的3’末端回折形成一个小引物末端,逆转录酶又以第一条cDNA链为模板再合成第二第cDNA链,至此,完成逆转录全过程,合成双链cDNA。
逆转录现在已成为一项重要的分子生物学技术,广泛用于基因的克隆和表达。从逆转录病毒中提取的逆转录酶已商品化,最常用的有AMV逆转录酶。利用真核Mrna3’末端存在一段聚腺苷酸尾,可以合成一段寡聚胸苷酸(oligo(dT))用作引物,在逆转录酶催化下合成互补于mRNA的cRNA链,然后再用RNase H将mRNA消化掉,再加入大肠杆菌的DNA聚合酶I催化合成另一条DNA链,即完成了从mRNA到双链DNA的逆转录过程。
所得到的双链cDNA分子经S1核酸酶切平两端后接一个有限制酶切点的接头(linker),再经特定的限制酶消化产生粘性末端,即可与含互补末端的载体进行连接。常用的克隆载体是λ噬菌体DNA,如λgt,EMBL和Charon系列等。用这类载体可以得到包含104以上的转化子的文库,再经前面介绍的筛选方法筛选特定基因克隆。用这种技术获得的DNA探针不含有内含子序列。因此尤其适用于基因表达的检测。
(三)RNA探针
RNA探针是一类很有前途的核酸探针,由于RNA是单链分子,所以它与靶序列的杂交反应效率极高。早期采用的RNA探针是细胞mRNA探针和病毒RNA探针,这些RNA是在细胞基因转录或病毒复制过程中得到标记的,标记效率往往不高,且受到多种因素的制约。这类RNA探针主要用于研究目的,而不是用于检测。例如,在筛选逆转录病毒人类免疫缺陷病毒(HIV)的基因组DNA克隆时,因无DNA探针可利用,就利用HIV的全套标记mRNA作为探针,成功地筛选到多株HIV基因组DNA克隆。又如进行中的转录分析(nuclear run on transcrip-tion assay)时,在体外将细胞核分离出来,然后在α-32P-ATP的存在下进行转录,所合成mR-NA均掺入同位素而得到标记,此混合mRNA与固定于硝酸纤维素滤膜上的某一特定的基因的DNA进行杂交,便可反映出该基因的转录状态,这是一种反向探针实验技术。
近几年体外转录技术不断完善,已相继建立了单向和双向体外转录系统。该系统主要基于一类新型载体pSP和pGEM,这类载体在多克隆位点两侧分别带有SP6启动子和T7启动子,在SP6RNA聚合酶或T7RNA聚合酶作用下可以进行RNA转录,如果在多克隆位点接头中插入了外源DNA片段,则可以此DNA两条链中的一条为模板转录生成RNA。这种体外转录反应效率很高,在1h内可合成近10μg的RNA产生,只要在底物中加入适量的放射性或生物素标记的NTP,则所合成的RNA可得到高效标记。该方法能有效地控制探针的长度并可提高标记物的利用率。
值得一提的是,通过改变外源基因的插入方向或选用不同的RNA聚合酶,可以控制RNA的转录方向,即以哪条DNA链以模板转录RNA。这种可以得到同义RNA探针(与mRNA同序列)和反义RNA探针(与mRNA互补),反义RNA又称cRNA,除可用于反义核酸研究外,还可用于检测mRNA的表达水平。在这种情况下,因为探针和靶序列均为单链,所以杂交的效率要比DNA-DNA杂交高几个数量级。RNA探针除可用于检测DNA和mRNA外,还有一个重要用途,在研究基因表达时,常常需要观察该基因的转录状况。在原核表达系统中外源基因不仅进行正向转录,有时还存在反向转录(即生成反义RNA),这种现象往往是外源基因表达不高的重要原因。另外,在真核系统,某些基因也存在反向转录,产生反义RNA,参与自身表达的调控。在这些情况下,要准确测定正向和反向转录水平就不能用双链DNA探针,而只能用RNA探针或单链DNA探针。
综上所述,RNA探针和cRNA探针具有DNA探针所不能比拟的高杂交效率,但RNA探针也存在易于降解和标记方法复杂等缺点。
(四)寡核酸探针
前述三种探针均是可克隆的,一般情况下,只要有克隆的探针,就不用寡核苷酸探针。在DNA序列未知而必须首先进行克隆以便绘制酶谱和测序时,也常应用克隆。克隆探针一般较寡核苷酸探针特异性强,复杂度也高,从统计学角度而言,较长的序列随机碰撞互补序列的机会较短序列少,克隆探针的另一优点是,可获得较强的杂交信号,因为克隆探针较寡核苷酸探针掺入的可检测标记基因更多。但是,较长的探针对于靶序列变异的识别能力又有所降低。对于仅是单个碱基或少数碱基不同的两序列,克隆探针不能区分,往往杂交信号相当。这既是其优点,又是其缺点。优点是当用于检测病原微生物时,不会因病毒或细菌DNA的少许变异而漏诊,缺点则是不能用于点突变的检测。这种情况下,通常要采用化学合成的寡核苷酸探针。
合成的寡核苷酸探针具有一些独特的优点:①由于链短,其序列复杂度低,分子量小,所以和等量靶位点完全杂交的时间比克隆探针短,如20nt的寡核苷酸探针在浓度为100ng/ml,靶序列为1~100pg、1kb片段或3×10-18~3×10-16mol/L时,达到最大程度的杂交只需10min,而用2kb的克隆探针在同样条件下达到完全杂交则需16h。②寡核苷酸探针可识别靶序列内1个碱基的变化,因为短探针中碱基的错配能大幅度地降低杂交体的Tm值。③一次可大量合成寡核苷酸探针(1~10mg),使得这种探针价格低廉,与克隆探针一样,寡核苷酸探针能够用酶学或化学方法修饰以进行非放射性标记物的标记。尽管克隆探针较特异,但通过细心筛选序列和/或选择相对长的序列(>30nt)亦可设计出非常特异的寡核苷酸探针。最常用的寡核苷酸探针有18~40个碱基,目前的合成仪可有效地合成至少50个碱基的探针。下面是筛选寡核苷酸针的一些原则。
①长18~50nt,较长探针杂交时间较长,合成量低;较短探针特异性会差些。
②碱基成分:G+C含量为40%~60%,超出此范围则会增加非特异杂交。
③探针分子内不应存在互补区,否则会出现抑制探针杂交的“发夹”状结构。
④避免单一碱基的重复出现(不能多于4个),如-CCCCC-。
⑤一旦选定某一序更符合上述标准,最好将序列与核酸库中核酸序列比较,探针序列应与含靶序列的核酸杂交,而与非靶区域的同源性不能超过70%或有连续8个或更多的碱基的同源,否则,该探针不能用。
按上述原则选出的探针会增加成功的机会,选定后进行合成与标记,并摸索合适的杂交条件。方法是制备几张点有特异靶DNA和不相关DNA的膜,各膜分别在不同温度下与探针杂交,特异靶DNA杂交信号强而非特异DNA不产生任何杂交反应的就是最适杂交温度。在进行点突变检测杂交的反应时,洗膜条件和温度物选择往往更为重要。所选漂洗条件必需使野生型靶DNA与探针产生强的杂交信号而突变型靶DNA则不产生杂交信号,这可以通过逐渐提高洗膜温度来完成。
寡核苷酸探针还有一个重要用途。在用于检测单个碱基差异时尚可采用一种称为寡核苷酸限制(oligonucleotide restriction)的技术。该技术只有在突变点位于某一限制性内切酶识别位点时才有效。例如,镰刀状红细胞贫血是因β珠蛋白基因的第6个寡码子由GAG变成GTG,从而导致所编码氨基酸由酪氨酸变成缬氨酸。突变的β-珠蛋白功能异常,称作S珠蛋白,而野生型称为A珠蛋白,其基因型分别为βS和βA。恰好突变点A→T位于Del I的识别序列CT-NAG之内,这就为设计寡核苷酸限制实验创造了条件。方法是合成一个长40个碱基的寡核苷酸探针,其5’末端距突变碱基有11个碱基,该探针与βA基因的非编码链互补。将此探针的5’末端标记上32P。杂交方法采用液相杂交法,即在液相中将靶DNA变性解链,然后与探针退火,产生杂交体。如靶DNA为βA型,则两条链完全互补,并产生Dde I的酶切位点;如待检DNA为βS型,则所形成的杂交体中两条链在突变碱基处不配对,从而不能被Del I所识别。用Del I消化杂交DNA,显然βA会被切开而βS不被切开。βADNA杂交体被切开后,5’端探针序列因只有8个碱基,与杂交链结合不紧而解离,从而产生游离的5’端标记8核苷酸单链。不被切开的βS杂交体尚可被另一个限制酶Hinf I消化,该酶的识别位点紧靠Del I 识别位点上游。βS杂交DNA经Hinf I消化后,将释出探针DNA的5’末端3核苷酸小片段。βADNA杂交体因已无Hinf I识别序列,故而不能被Hinf I消化。这样βA和βSDNA经此寡核苷酸探针杂交和Del I及Hinf I消化后,分别产生游离的8核苷酸(8nt)和3核苷酸(3nt) 片段,它们可以经电泳分离后进行放射自显影而获证实。藉此策略,可轻易将各种β珠蛋白突变型鉴别开,如纯合野生型AA结果为仅有8nt片段,纯合突变型SS则仅可检出3nt片段,而杂合子AS型则两种片段均存在。
四、核酸探针的标记和检测(详见每十九章 )
分子杂交是核酸链间碱基配对规则的一种结合方式,是核酸的重要理化特性。利用分子杂交这一特性来对特定核酸序列进行检测,必须将杂交链中的一条用某种可以检测的分子进行标记,这条链就称为核酸探针。因此,核酸探针的制备是分子杂交技术的关键。最早采用的也是目前最常用的核酸探针标记方法是放射性同位素标记。常用的放射性同位素有32P和35S前者能量高,信号强,最常用。放射性同位素标记探针虽然敏感度高,但却存在辐射危害和半衰期限制(32P半衰期为14.3天,35S半衰期为87.1天,125I的半衰期为60天),3H的半衰期长达12.3年,但它所释放β放射线能量太低(0.018Mev),只能用于组织原位杂交。由于同位素标记的探针在使用过程中存在着上述缺点,近些年来,人们在寻找非航船性标记物方面取得了很大进展,国际上已有多家公司相继推出多种非放射性探针标记试剂盒,在国内也已具备生物素类标记物的生产能力,并有相应试剂出售。目前,非放射性标记物有下述几类:金属如Hg,荧光物质如FITC;半抗原如地高辛;生物素;酶类如辣根过氧化物酶(HRP)。半乳糖苷酶或碱性磷酸酶(AKP)等。不同的标记物,所标记探针的方法及检测方法也各异。下面仅就国际上较常用的,有实用价值或发展前景的几种核酸标记方法及其显示方法分两方面简述如下。
(一)核酸探针的常用酶促标记技术
1.缺口平移 该技术由Kelly等于1970年创立。其原理是首先用DNA酶在双链DNA探针分子的一条链上制造一些缺口(nick ),缺口处会形成3’—羟基末端,这时再在大肠杆菌DNA聚合酶I的催化下将核苷酸残基加在3’-羟基上,同时,根据大肠杆菌酶DNA聚合酶I的5’→3’核酸外切酶活性,此酶将缺口5’侧核苷酸依次切除。其结果是在缺口平移(nick tr-anslation)。根据这个原理,如果用高强度的放射性核苷酸(通常为α-32PdATP)置换先前存在的核苷酸,则可制备比活性高达108cpm(每分钟计数)/μg的32P标记DNA。用缺口平移法标记的DNA探针能满足大多数杂交要求。
2.DNA快速末端标记 大肠杆菌DNA聚合酶i 经枯草杆菌蛋白酶切割可得到两条多肽链,其中分子量为76kd的大片段称为Klenow片段。该酶具有完整聚合酶I的5’→3’聚合酶活性和3’→5’核酸外切酶活性,但缺乏5’→3’核酸外切酶活性。利用Klenow片段可以填补由限制酶消解DNA所产生的3’凹陷末端。因此,用这种方法可以标记双链DNA的凹陷3’末端。用Klenow片段标记末端一般只用一种[α-32P]dNTP,加入反应的[α-32P]dNTP取决于DNA末端延伸的5’末端序列,例如,用EcorI切割DNA所产生的末端用[α-32P]dNTP标记。标记反应可在一种限制酶消解DNA后立即进行,不需去除限制酶或使其失活,也不需更换缓冲液,具有3’延伸的DNA末端不能被Klenow片段有效在标记,欲标记这类分子可用T4DNA聚合酶。
选用这种标记方法是为了产生可用于凝胶电泳时作大小参照物的DNA片段。因为标记的DNA片段与其摩尔浓度成比例,而不与片段大小成比例,在限制酶消化物中,小的和大的片段都以相同程度被标记。因此,可使用放射自显影术确定不有被溴化乙锭染色所观察到的大小DNA带,尤其适用于Southern吸印杂交时分子量标志物的标记。通过选择相应标记的dNTP,该法还可以只标记DNA分子的一端。例如,若DNA片段的两个末端分别是Bam H I和Hind III 粘膜端,在反应中只加入[α-32P]dNTP或[α-32P]dGTP,使可选择性标记两末端之一。
3.用T4多核苷酸激酶标记DNa 5’末端 寡核苷酸探针或短的RNA和DNA探针可选用此法标记,寡核苷酸探针一般多用这种标记。T4多核苷酸激酶(polynucleotide kinase, PNK)是由T4噬菌体感染的大肠杆菌中提取的,此酶能催化ATP的γ-磷酸转移至DNA或RNA的5’-OH末端。在过量ADP存在时,也可促进磷酸交换反应,使PNK将DNA末端5’磷酸转移到ADP上生成ATP,然后催化[α-32P]dNTP上的标记磷酸转移至DNA的5’末端,从而使DNA重新磷酸化,并藉此得到标记。显然,PNK标记DNA末端需要[γ-32P]dNTP,这与前述酶促标记方法不同。通常,对于5’磷酸化的DNA探针,要先用碱性磷酸酶去掉磷酸基团,然后再用于PNK催化的5’末端标记,这样标记效率较高。
4.随机引物延伸 这是以单链DNA或RNA模板合成高比活性32P标记探针所选用的方法。原理是使长6~8nt的寡核苷酸片段与变性的DNA或RNA模板退火,在DNA聚合酶I或反转录酶的作用下,以每一个退火到模板上的寡核苷酸片段为引物引发DNA链的合成,在反应时将[α-32P]dNTP掺入合成链,即得到标记。变性处理后,新合成链(探针片段)与模板解离,即得到无数各种大小的探针DNA。因为所用寡核苷酸片段很短,在低温条件下可与模板DNA随机发生退火反应,因此被称为随机引物(random primer)。这种随机引物可用小牛胸腺DNA或鱼精DNA制备。
用随机引物法标记的DNA探针或cDNA探针比活性显著高于缺口平移法,且结果较为稳定。这种方法尤其适用于真核DNA探针,因为随机引物来自真核DNA,其与真核序列的退火率要高于原核序列。因此,对于克隆的DAN探针,常先将插入探针DNA切下来回收后再标记,而缺口平移法可直接用于全质粒的标记。
5.聚合酶链反应(详见第二十二章) 聚合酶链反应(polymerase chain reaction, PCR)是一种分子生物学新技术,由美国Cetus公司人类遗传学部的Kary.B. Mullis于1985年创立。该技术利用两个与相反链杂交并随着于靶DNA两侧的寡核苷酸引物经酶促合成特异的DNA片段,包括模板变性,引物退火和引物延伸三个步骤的反复循环,最终两引物所夹靶DNA得到千万倍以上的扩增。因此,PCR技术已成为一项极为有价值的技术并已迅速推广应用。
PCR技术有许多重要用途,其中之一便是可用来标记高比活性DNA探针。PCR技术具有很高的特异性,可在1~2h之内在量合成探针DNA片段,如果在底物中加入[α-32P]dNTP或其它标记的dNTP,则探针DNA合成过程中可得到很好的标记,标记物的掺入率可高达70%~80%。因此,PCR标记技术特别适用于大规模检测和非放射性标记。该法的缺点是要合成一对特异性PCR引物。使用从探针DNA上制备的小片段作引物也能取得较好的标记效果。
(二)核酸探针的非放射性标记技术
1.光敏生物素标记核酸 目前使用的光标生物素试剂有两种:光生物素(乙酸盐)和补骨脂素生物素。它们都是由一个光敏基团、一个连接臂和一个生物素基团组成。光生物素的光敏基团是-N3,它在光作用下可与核酸中的碱基结合。补骨脂素生物素中的光敏基团补骨脂素在光照(320~400μm)下,可与单链或双链核酸发生反应,反应主要在T上,C上也有一定程度的反应。光敏生物素的连接臂含6~12个碳原子,用以减少探针杂交时的空间位阻。光敏生物素标记核酸,方法简单,灵敏度也可达到pg水平,可用于外源基因的检测。最近出现了一种新的光敏活性DNA生物素试剂,即生物素-聚乙二醇-当归素(BPA)。BPA的DNA结合部分是一个活性糖香豆素衍生物,在长波UV下它可与DNA碱基共价键结合。BPA反应物与DNA结合比光敏生物素更特异,在可见光下它不与核酸反应,这个特性可使BPA只标记粗制细胞裂解物中的核酸,而不标记蛋白、多糖和其他细胞大分子。
2.酶促生物素标记核酸 以生物素化的脱氧核苷三磷酸(Bio-11-dUTP,Bio –7 – dATP、Bio-11-dCTP)等代替相应32P标记的脱氧核苷三磷酸,经DNA聚合酶作用掺入DNA。Bio-dUTP代替dTTP,Bio-dATP代替dATP,Bio- dCTP代替dCTP。Bio –11-dUTP的11是指生物素基团与脱氧核苷酸之间连接臂的碳链长度。常用的酶促生物素标记DNA的方法有缺口平移法和随机引物延伸法。
3.寡核苷酸的生物素末端标记 有5’-磷酸的化学标记法和3’-OH的酶促标记法。前者将寡核苷酸的5’-磷酸接上一个乙二胺,然后用琥珀酰亚胺生物素,将生物素基团连接在磷酸酰胺基上。后者是用末端转移将Bio-11–dUTP加于其3’-OH端(脱去一个焦磷酸)。
4.酶标DNA 标记试剂是辣根过氧化酶(HRP)或碱性磷酸酶(AP)。通过对苯醌(PBQ)与聚乙烯亚胺(PEI)连接而成(HRP-PBQ-PEI+),此试剂在戊二醛的作用下与变性的DNA结合,使HRP与DNA连接在一起,组成HRP标记的DNA探针。
5.酶标寡核苷酸 包括核苷酸5’末端标记HRP法和内部标记AP法。前者是在HRP中产生一个-HS反应基团,在寡核苷酸合成终了加在5’端,带一个C6的-HS基,与活化的HRP反应生成5’-HRP寡核苷酸。后者是在全成寡核苷酸过程中将一个5’带连接臂及CF3基团的尿苷3’亚磷酰亚胺合成在寡核苷酸链中,合成后此活化的寡核苷酸与AP反应即得到AP标记的寡核苷酸。
6.DNA半抗原标记 其原理与Bio-11-dTUP相同,只是用毛地黄甙代替生物素形成Dig –11- dUTP,酶促掺入DNA分子。用抗毛地黄甙抗体检测标记在DNA上的半抗原分子Digoxigenin(地高辛)
(三)非放射性探针的显示体系
1.AP显色体系
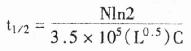
BCI –OH+NBT→紫色↓
ASO:等位基因特异的寡核苷酸,BCIP:5溴-4氯-3吲哚磷酸,NBT:四氮唑蓝,Pi:磷酸。
2.HRP显色体系
HRP + H2O2[HRP·H2O2]
ODA –NH2 +[HRP ·H2O2]枣→ODA-N = ODA/棕色↓+HRP +H2O
ODA:邻-联茴香胺。
3.ABC显色体系
DNA –B+ SA – AP→DNa – B – SA或DNa – B +SA - BAP→DNa –B –SA –BAP
经上述两反应,把AP连接在DNA上以后,再进行AP酶显色。这里SA为Streptavidin(链酶亲合素),BAP为生物素化的磷酸酶,B为生物素(biotin),ABC为Avidin – Biotin - Enzyme com-plex, 即亲合素- 生物素-酶复合物。
以上反应AP亦可用HRP代替。
表18-3曱核酸探针的标记分子
| 标记物性质 | 标记分子 | 标记方法 | 杂交体的检测法 |
| 放射性分子 | [α-32P]dNTP | NT、PCR、RPI | 放射自显影或计数 |
| [γ-32P]dNTP | TL | 放射自显影或计数 | |
| 125I | 碘化 | 放射自显影或计数 | |
| 35S | NT | 放射自显影或计数 | |
| 3H | NT | 放射自显影或计数 | |
| 非放射性分子 | |||
| 生物素 | Bio-11-dUTP | NT、TL、PCR | 酶标亲合素或酶标抗生物素抗体显色 |
| 光敏生物素 | 600W 可见光照 | 同上 | |
| 生物素化补骨脂素 | 365nm紫外线照 | 同上 | |
| 生物素酰-e-氨基乙酸 | 化学合成法 | 同上 | |
| N-羟基丁二酰亚胺脂 | 酶标亲合素或酶标抗生物素抗体显色 | ||
| 生物素肼 | 化学合成法 | 酶标亲合素或酶标抗生物素抗体显色 | |
| 酶 | 微过氧化物酶 | 直接法或合成法 | 直接底物显色或用酶标抗体+ 底物显色 |
| 碱性磷酸酯酶 | 直接法或合成法 | 同上 | |
| 荧光素 | 罗丹明和FITC | 合成法 | 直接荧光显微镜观察或酶标抗体+ 底物显色 |
| 化学发光标记物 | 化学发光测量或自显影 | ||
| 抗双链单抗 | 酶标或荧光标记单抗 | 化学法 | 直接底物显色 |
| 稀有金属 | Eu3- | 化学法 | 时间分辨荧光 |
| 重金属 | Ag | 化学法 | |
| Hg | 化学法 | ||
| 半抗原 | 磺酸胞嘧啶 | 化学法 | 酶标单抗 + 底物显色 |
| 地高辛 | RPL | 酶标抗体 + 底物显色 |
* :NT:缺口平移; PCR:聚合酶链反应;RPL: 随机引物标记; TL:末端标记
4.非放射发光自显影若将AP或HRP的显色底物根据光化学原理换成一种酶解后产生光子的化合物,可用自显影技术暴光X线片显示。
(1)HRP发光自显影:氨基苯-甲酰肼在HRP与H2O2的作用下氧化为氨基苯二甲酸,同时放出N2及发光(波长428nm)。发光时加入某些酚的衍生物时可增强发光上千倍。反应如下图:
(2)AP的发光自显影:AP的发光底物是金刚烷二氧丁环磷酸盐(AMPPD),它含有磷酸酯键在AP的作用下水解下一个磷酸,进而由分子内过氧键提供能源分解产生金刚酮和激发态的甲基间-氧苯甲酸阴离子,当此阴离子恢复到基态时发出光子。可用波拉黑白片(621型)直接暴光显影。显影信号强度比BCIP/NBP显色法强两上数量级。是很前景的显示体系。
其发光反应的原理如下:
HRP的发光原理:
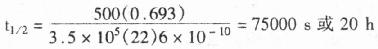
AP的发光原理:
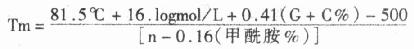
五、核酸分子杂交的类型
随着基因工程研究技术的迅猛发展,新的核酸分子杂交类型和方法在不断涌现和完善。核酸分子杂交可按作用环境大致分为固相杂交和液相杂交两种类型。固相杂交是将参加反应的一条核酸链先固定在固体支持物上,一条反应核酸游离在溶液中。固体支持物有硝酸纤维素滤膜、尼龙膜、乳胶颗粒、磁珠和微孔板等。液相杂交所参加反应的两条核酸链都游离在溶液中。
由于固相杂交后,未杂交的游离片段可容易地漂洗除去,膜上留下的杂交物容易检测和能防止靶DNA自我复性等优点,故该法最为常用。常用的固相杂交类型有:菌落原位杂交、斑点杂交、狭缝杂交、Southern印迹杂交、Northern印迹杂交、组织原位杂交和夹心杂交等。
液相杂交是一种研究最早且操作复合的杂交类型,在过去的30年里虽有时被应用,但总不如固相杂交那样普遍。其主要原因是杂交后过量的未杂交探针在溶液中除去较为困难和误差较高。近几年由于杂交检测技术的不断改进,商业性基因探针诊断盒的实际应用,推动了液相杂交技术的迅速发展。下面对固相杂交和液相杂交分别进行介绍。
(一)固相膜核酸分子杂交方法
固相核酸杂交多是在膜上进行,因此,以下主要介绍固相膜的核酸分子杂交方法:
1.DNA的变性 DNA变性解链是杂交成功的关键。Southern印迹杂交时DNA在凝胶中变性,变性方法是将凝胶浸在数倍体积的1.5mol/l NaCl和0.5mol/l NaOH中1h然后用数倍体积的1mol/l Tris –HCl(Ph8.0)和1.5mol/l NaCl溶液中和1h。DNA受酸、碱、热等处理均能发生变性,但强酸会使核酸降解。一般认为碱变性较好,可避免DNA降解。热变性在要低DNA浓度(100μg/ml)和低盐浓度(0.1mol/l SSC 含15mmol/l NaCl - 1.5mmol/L柠檬酸三钠,pH7.0)下进行。用SSC稀释DNA溶液为50μg/ml,加10mol/l NaOH使最终浓度为0.1mol/L(pH约12.8),室温变性10min,很快置冰盐水中,用10min/l HCl或5mol/l NaH2PO4调pH到7~8[亦可用碱变性后,调至中性,再加热100。c 10min],DNA变怀可用OD260增加(约30%~40%)来检测,变性DNA醇沉淀呈雪样,完全失去纤维状沉淀。变性后加入等量冷的12×SSC,冰浴保存。
2.变性DNA在硝酸纤维素膜上的固定 硝酸纤维素滤膜(孔径0.45μm)先在蒸馏水中充分浸泡,再用6×SSC浸泡30min~2h,凉干。DNA样品转移或加至硝酸纤维素膜上后,先室温干燥4h,然后在80。C真空干燥2h。
3.预杂交 湿润的滤膜放入可加热封口的塑料袋中,按每平方厘米滤膜加0.2ml预热至60。C的预杂交液(6×SSC,0.5%,SDS,5×Denhardt液,100μg/ml鲑鱼精DNA)。鲑鱼精DNA需经过剪切和DNA酶消化处理,然后酒精沉淀纯化,调浓度至10mg/ml,用前放100。C水浴中煮沸变性10min,冰水骤冷。尽可能将袋中气泡赶尽,可封口器将袋口封住。将杂交袋浸入68。C水浴保温3~12h。当预杂交液温度升至68。C时,在滤膜表面常会形成小气泡。轻轻晃动袋中液体即可除去这些小气泡,这一点对于保证滤膜表面充分浸润预杂交液很重要。
4.杂交 从水浴中取出塑料袋,用剪刀剪开一角,尽可能挤净预杂交液。用吸管或大枪头将杂交液加入袋中,用恰好是足量的液体保持滤膜湿润(50μl/cm2)。溶液的组成是6×SSC,0.01mol/l EDTA, 变性的标记核酸探针,5×Denhardt液,0.5%SDS, 100μg/ml变性的鲑鱼精DNA。尽可能赶尽气泡后,将塑料袋严密封口,杂交反应在68℃水浴中进行,所需时间视探针和检测靶DNA的性质及探针的比活性等情况而定,一般4~20h。
5.洗膜 取出塑料袋,用剪刀剪开,小心取出滤膜,立即浸入盛有2×SSC和0.5%SDS溶液的盘中,室温下漂洗5min。再将滤膜移入2×SSC和0.1%SDS溶液中,室温下洗涤15min(轻轻摇动)。然后将滤膜移入0.1×SSC和0.5%SDS溶液中;68℃轻轻摇动保温2h,更换缓冲液后继续保温30min。
洗膜的温度一般应控制在低于Tm值12℃以上。(Tm = 69.3 + 0.41x(G +C) %)。双链DNA的Tm值随错配碱基对数每增加1%而递减1℃。
6.结果显示 非放射性检测方法前已述及,此处主要介绍放射性测定方法。固相膜的放射性杂交结果显示有两种方式,一是放射性自显影法,另一种是液闪计数法。前一种方法比较简单,只需将杂交膜与X光片在暗盒中曝光数小时至数天,再显影、定影即可。对于杂交信号较弱的固相膜,用一块增感屏可显著增强曝光强度。此外,为了减弱32P的散射,曝光通常在-20℃或-80℃下进行。液闪计数法主要用于打点和狭缝杂交及为了比较两个杂交信号的强弱等情形。方法是将完成杂交的膜在漂洗结束后剪成小块(每份样品1块),80℃真空干燥后装闪烁瓶。加入2~5ml闪烁液,剪2~3块无样品膜块作为本底对照。在液体闪烁计数器上自动计数。液体计数测定放射性强度也可在放射自显影之后进行。
(二)固相核酸分子杂交类型
1.菌落原位杂交(Colonyin situ hybridization) 菌落原位杂交是将细菌从培养平板转移到硝酸纤维素滤膜上,然后将滤膜上的菌落裂菌以释出DNA。将NDA烘干固定于膜上与32P标记的探针杂交,放射自显影检测菌落杂交信号,并与平板上的菌落对位。
实验步骤如下:
①将硝酸纤维素滤膜置于含抗生素的平皿琼脂培养基上,用无菌牙签挑取单菌落种于滤膜和主琼脂平板上,排列成方格栅,膜和板上菌落位置相同。
②培养细菌至产生1~2mm大小的菌落。
③在一块平皿中置4张滤纸,用10%SDS浸透,倒掉多余液体。将带有菌落的滤膜取下,轻轻置于滤纸上,菌落面上在,注意防止在滤膜底面存有气泡。
④5min后,将滤膜转至用变性溶液(0.5mol/l NaCl , 0.5mol/L Tris·NaCl)浸湿的滤纸上,放置10min。
⑤将滤膜转至中和溶液(1.5mol/lNaCl , 0.5mol/L Tris·HCl ,pH8.0)浸湿的滤纸上,放置10min。重复中和1次。
⑥将滤膜移至用2×SSPE溶液浸过的滤纸上,放置10min。SSPE配成20×贮备液:3.6mol/l NaCl, 0.2mol/L NaH2PO4(PH7.4), 20mmol/L EDTA(pH7.4)。
⑦将滤膜用滤纸吸干,80℃真空烘干2h。
以下操作参考前述。
2.斑点杂交(Dot blot)斑点杂交法是将被检标本点到膜上,烘烤固定。这种方法耗时短,可做半定量分析,一张膜上可同时检测多个样品。为使点样准确方便,市售有多种多管吸印仪(manifolds),如Minifold I和II、Bio-Dot (Bio –Rad )和Hybri –Dot,它们有许多孔,样品加到孔中,在负压下就会流到膜上呈斑点状或狭缝状,反复冲洗进样孔,取出膜烤干或紫外线照射以固定标本,这时的膜就可以进行杂交。
(1)DAN斑点杂交
①先将膜在水中浸湿,再放到15×SSC中。
②将DNA样品溶于水或TE,煮沸5min,冰中速冷。
③用铅笔在滤膜上标好位置,将DNA点样于膜上。每个样品一般点50μl(2~10μg DNA)。
④将膜烘干,密封保存备用。
(2)RNA斑点杂交:与上法类似,每个样品至多加10μg总RNA(经酚/氯仿或异硫氰酸胍提取纯化)。方法是将RNA溶于5μlDEPC水,加15μl甲醛/SSC缓冲液(10×SSC中含0.15mol/l甲醛)使RNA变性。然后取5~8μl点样于处理好的滤膜上,烘干。
培养细胞,标本处理技术可以简化,不用提取和纯化RNA。方法是用含0.5%Nonidet P40的低渗缓冲液对多种动物细胞作简单处理,离心去掉细胞核和细胞碎片,就得到基本不带DNA而富含RNA的细胞质提取物,这一粗RNA在高盐下用甲醛变性,不需加工直接点到硝酸纤维素膜上。本法可以快速检测大量标本,而只需极少量的细胞(5×104)或组织。
整个RNA实验中,要防止激活内源性RNase,有许多种预防措施,有一种是在样品中加入核糖核苷氧矾基复合物(RVC)。
(3)完整细胞斑点杂交:应用类似检测细菌菌落的方法,可以对细胞培养物的特异序列进行快速检测。将整个细胞点到膜上,经NaOH处理,使DNA暴露、变性和固定,再按常规方法进行杂交与检测。有人曾用此法从105个培养细胞中检测到少至5pg 的Epstein– Barr病毒DNA。完整细胞斑点印迹法可以用于筛选大量标本,因为它是使细胞直接在膜上溶解,所以DNA含量甚至比常用的提取法还高,又不影响与32P标记的探针杂交。但它不适于非放射性标记探针,因为DNA纯度不够,会产生高本底。
3.Southern印迹杂交(southern blot )Southern blot 是研究DNA图谱的基本技术,在遗传诊断DNA图谱分析及PCR产物分析等方面有重要价值。Southern印迹杂交基本方法是将DNA标本用限制性内切酶消化后,经琼脂糖凝胶电泳分离各酶解片段,然后经碱变性,Tris缓冲液中和,高盐下通过毛吸作用将DNA从凝胶中转印至硝酸纤维素滤膜上,烘干固定后即可用于杂交。凝胶中DNA片段的相对位置在DNA片段转移到滤膜的过程中继续保持着。附着在滤膜上的DNA与32P标记的探针杂交,利用放射自显影术确定探针互补的每条DNA带的位置,从而可以确定在众多酶解产物中含某一特定序列的DNA片段的位置和大小。
(1)琼脂糖凝胶电泳:利用琼脂糖凝胶电泳可以很容易地将DNA限制酶消化片段(0.3~25kb)分离开。分离大分子DNA片段(800~12000bp)用低浓度琼脂糖(0.7%),分离小分子片段(500~1000bp)用高浓度琼脂糖(1.0%),300~5000bp的片段则用1.3%的琼脂糖凝胶,根据分离样品量、分离速度和分辨率要求的不同,可选用不同规格的电泳槽。
电泳时,同时将标记物加到旁边孔中,便于确定样品DNA的分子量。20伏恒压电泳过夜。电泳毕,将胶浸到含0.5μg/ml EB的TBE缓冲液中染色30min,也可将EB直接加到电泳缓冲液中或在配胶前加入胶中,在254nm短波透射灯下拍照,加橙黄色滤色镜,使用高速一次成像胶片,光圈f4.5,曝光20~40s。
(2)硝酸纤维素滤膜吸印。
①将胶切成合适大小,切去右上角作为记号。
②将胶放进盛有变性缓冲液(1.5mol/lNaCl, 0.5mol/L NaOH)的盘中轻摇动15min。
③换到中和缓冲液(1mol/LTris·HCl , pH8.0, 1.5mol/L NaCl)中轻摇动30min。
④裁一张硝酸纤维素膜,2~4张3MM滤纸和一些吸印纸(可用卫生纸),都与胶的大小相同(硝酸纤维素膜和吸印纸不能比胶大,否则易形成旁路),先将硝酸纤维素膜浸到水中,再放入10×SSC中,接触胶和硝酸纤维素膜时都要戴橡胶手套操作。
⑤平盘上放一块比胶大的平板(盛胶槽翻过来即可),上面铺一张3MM滤纸,起灯芯作用,盘中加少量10×SSC缓冲液(2.5cm厚),不能没过平板,使3MM滤纸充分饱和。
⑥将胶倒扣到3MM滤纸上。
⑦浸湿的硝酸纤维素铺在胶上,对齐,铺膜时从一边逐渐放下,防止产生气泡,有气泡时,可用吸管赶出,不能让膜与胶下的滤纸直接接触。
⑧膜上放一张3MM滤纸,不能与胶接触。
⑨上面加吸印纸及重物(500g左右)。
⑩通过滤纸的灶芯作用,平盘中的缓冲液就会通过胶上移,从而将DNA吸印到膜上,及时更换浸湿的吸印纸。在室温下转印过夜。
⑾去除上面的东西,用镊了将膜取出,在6×SSC中洗一下(也可不洗)。
⑿自然干燥,80℃烤2h。
⒀这时的膜就可进行杂交,或室温密封保存。
4.Northern印迹杂交(Northern blot) 这是一种将RNA从琼脂糖凝胶中转印到硝酸纤维素膜上的方法。DNA印迹技术由Southern于1975年创建,称为Southern印迹技术。RNA印迹技术正好与DNA相对应,故被趣称为Northern印迹杂交,与此原理相似的蛋白质印迹技术则被称为western blot。Northern印迹杂交的RNA吸印与Southern印迹杂交的DNA吸印方法类似,只是在进样前用甲基氧化汞、乙二醛或甲醛使RNA变性,而不用NaOH,因为它会水解RNA的2’-羟基基团。RNA变性后有利于在转印过程中与硝酸纤维素膜结合,它同样可在高盐中进行转印,但在烘烤前与膜结合得并不牢固,所以在转印后不能用低盐缓冲液洗膜,否则RNA会被洗脱。在胶中不能加EB,因它为影响RNA与硝酸纤维素膜的结合,为测定片段大小,可在同一块胶上加标记物一同电泳,之后将标记物胶切下,上色、照像。样品胶则进行Northern转印,标记物胶上色的方法是在暗室中将其浸在含5μg/ml EB的0.1mol/L 醋酸铵中10min,在水中就可脱色。在紫外光下用一次成像相机拍照时,上色的RNA胶要尽可能少接触紫外光,若接触太多或白炽灯下暴露过久,会使RNA信号降低。从琼脂糖凝胶中分离功能完整的mR-NA时,甲基氢氧化汞是一种强力、可逆变性剂,但是有毒,因而许多人喜用甲醛作为变性剂。所有操作均应避免RNase的污染。
下面介绍RNA甲醛凝胶电泳和吸印方法:
(1)试剂
10×MSE缓冲液:0.2mol/L 吗啉代丙烷磺酸(MOPS),pH7.0, 50mmol/L 醋酸钠,1mmol/l EDTA, pH8.0。
5×样品缓冲液:50%甘油,1mmol/l EDTA, 0.4%溴酚蓝。
甲醛:用水配成37%浓度(12.3mol/L)。应在通风柜中操作,pH高于4.0。
去离子甲酰胺。
50mmol/L NaOH(含10mmol/l NaCl)。
0.1mol/L Tris·HCl,Ph7.5。
(2)步骤
①40ml水中加7g琼脂糖,煮沸溶解,冷却到60℃,加7ml 10×MSE缓冲液、11.5ml甲醛,加水定容 至70ml,混匀后 倒入盛胶槽。
②等胶凝固后,去掉梳子和胶布,将盛胶槽放入1×MSE缓冲液的电泳槽。
③使RNA变性(最多20μg):RNa 4.5μl,10×MSE缓冲液2.0μl,甲醛3.5μl,去离子甲酰胺10.0μl。
④55℃加热15min,冰浴冷却。
⑤加2μl 5×载样缓冲液。
⑥上样,同时加RNA标记物。
⑦60伏电泳过夜。
⑧取出凝胶,水中浸泡2次,每次5min。
⑨室温下将胶浸到50mmol/LNaOH和10mmol/l NaCl中45min,水解高分子RNA,以增强转印。
⑩室温下将胶浸到0.1mol/LTris·HCl (Ph7.5)中45min,使胶中和。
⑾20×SSC洗胶1h。
⑿20×SSC中过夜,转印到硝酸纤维素膜上。
⒀取出硝酸纤维膜,80℃真空烘烤2h。
5.组织原位杂交(Tissuein situ hybridization) 组织原位杂交简称原位杂交,指组织或细胞的原位杂交,它与菌落的原位杂交不同。菌落原位杂交需裂解细菌释出DNA,然后进行杂交。而原位杂交是经适当处理后,使细胞通透性增加,让探针进入细胞内与DNA或RNA杂交。因此原位杂交可以确定探针的互补序列在胞内的空间位置,这一点具有重要的生物学和病理学意义。例如,对致密染色体DNA的原位杂交可用于显示特定的序列的位置;对分裂期间核DNA的杂交可研究特定序列在染色质内的功能排布;与细胞RNA的杂交可精确分析任何一种RNA在细胞中和组织中的分布。此外,原位杂交还是显示细胞亚群分布和动向及病原微生物存在方式和部位的一种重要技术。
用于原位杂交的探针可以是单链或双链DNA,也可以是RNA探针。通常探针的长度以100~400nt为宜,过长则杂交效率减低。最近研究结果表明,寡核苷酸探针(16~30nt)能自由出入细菌和组织细胞壁,杂交效率明显高于长探针。因此,寡核苷酸探针和不对称PCR标记的小DNA探针或体外转录标记的RNA探针是组织原位杂交的优选探针。
探针的标记物可以是放射性同位素,也可以是非放射性生物素和半抗原等。放射性同位素中,3H和35S最为常用。3H标记的探针半衰期长,成像分辨率高,便于定位,缺点是能量低。35S标记探针活性较高,影像分辨率也较好。而32P能量过高,致使产生的影像模糊,不利于确定杂交位点。
原位杂交中,标本的固定条件是影响杂交效率的重要因素,标本组织蛋白质的消化程度对探针进入细胞极为重要。去除蛋白的方法是,用0.2mol/l HCl处理载玻片,用蛋白酶K消化,然后用不同浓度的乙醇脱水,原位杂交还是一种新技术,发展很快,在敏感性、特异性和稳定性上还需要进一步完善和提高(详见二十章 )。
6.固相夹心杂交Dunn等最早介绍了夹心杂交类型,Ranki等又作了进一步的改进。夹心杂交法比直接滤膜杂交法有两个主要的优点:①样品不需要固定,对粗制样品能做出可靠的检测;②用夹心杂交法比直接滤膜杂交法特异性强,因为只有两个杂交物都杂交才能产生可检测的信号。
固相夹心杂交需要两个靠近而不互相重叠的探针,一个作固相吸附探针,另一个作标记检测探针。样品基因组内核酸只有使这两个探针紧密相连才能形成夹心结构。需注意的是两个探针必须分别亚克隆进入两个分离的非同源载体内,以避免产生高的本底信号(如一个克隆人Puc19,另一克隆人pBR322)。
夹心杂交法可用滤膜和小珠固定吸附探针,使用小珠可更好地进行标准化试验和更容易对小量样品进行操作。Dahlen 等利用微孔板进行夹心杂交,可同时进行大量样品检测,他们先吸取DNA探针加到凹板中,然后用紫外线照射使其固定到塑料板上。用微孔板进行夹心杂交还可直接用于PCR技术。应用光敏生物标记探针检测PCR产物的敏感性和用32P标记探针(3×108cpm/μg)作16h放射自显影的Southern杂交的敏感性一样。用微孔板杂交的其它优点还包括同时做多份样品,加样、漂洗和读结果等步骤可以自动化。
7.其它杂交类型
(1)固化探针杂交:该法较少使用,原理是使未标记固化探针通过杂交与靶RNA或DNA结合,漂洗后,用酶标抗DNA:RNA抗体或抗DNA:DNA抗体与杂交物结合。将乳胶颗粒收集,吸附到膜上后漂洗,加入底物显色并进行测定。探针浓度2μg/ml,80℃杂交,可在10~15min完成,检测的敏感性为5×108靶序列。
(2)反向杂交:这个杂交类型是用标记的样品核酸与未标记的固化探针DNA杂交,故称为“反向杂交”。这种杂交方法的优点是在一次杂交反应中,可同时检测样品中几种核酸。这种杂交方式主要用于进行中的核酸转录试验和多种病原微生物的检测。前者是在转录过程中标记RNA探针,后者可用光敏生物素制剂BPA标记样品核酸。
(三)固相膜核酸杂交的几点说明
1.杂交膜的选用杂交膜是一种多孔、表面积很大的固相载体,核酸一旦固定在上面,就可用杂交法进行检测。最常使用的膜是硝酸纤维素膜,用于放射性和非放射笥标记探针都很方便,产生的本底浅,与核酸结合的化学性质不是很清楚,推测为非共价键结合。经80℃烤干2h和杂交处理后,核酸仍不会脱落。硝酸纤维素膜的另一特点是只与蛋白有微弱非特异结合,这在使用非同位素探针中尤为有用。硝酸纤维素膜的缺点是结合核酸能力的大小取决于转印条件和高浓度盐(>10×SSC),与小片段核酸(<200bp)结合不牢,质地脆,不易操作。
尼龙膜在某些方面比硝酸纤维素膜好,它的强度大、耐用,可与小至10bp的片段共价结合。在低离子浓度缓冲液等多种条件下,它们都可与DNA单链或RNA紧密结合,且多数膜不需烧烤。尼龙膜韧性好,可反复处理与杂交,而不丢失被检标本。它通过疏水键和离子键与核酸结合,结合力为350~500μg/cm2,比硝酸纤维素膜(80~100μg/cm2)强许多。尼龙膜的缺点是对蛋白有高亲合力,不宜使用非同位素探针。
2.“噪音”的排除“噪音”(noise)是固相膜杂交方法常遇到的问题,指标记DNA结合到空白膜上的放射性计数,即本底。这个问题的克服一是使用高纯度的核酸制品和充分严格的杂交条件:二是选择合适的杂交反应液和对膜进行处理。研究发现,随着离子强度的增加,空白膜(未固定DNA对照膜)上的“噪音”水平增加,而在50%甲酚胺6×SSC的杂交反应液中充分封闭膜上的多余非特异结合位点。
(四)液相核酸分子杂交类型
1.
(1)HAP吸附杂交:羟基磷灰石(HAP)层析或吸附是液相杂交中最早使用的方法。在液相中杂交后,DNA:DNA杂交双链在低盐条件可特异地吸附到HAP上。通过离心使吸附有核酸双链的HAP沉淀,再用缓冲液离心漂洗几次HAP,然后将HAP置于计数器上进行放射性计数。
(2)亲合吸附杂交:生物素标记DNA探针与溶液中过量的靶RNA杂交,杂交物吸附到酰化亲合素包被的固相支持物(如小球)上,用特异性抗DNA:RNA杂交物的酶标单克隆抗体与固相支持物上的杂交物反应,加入酶显色底物,这个系统可快速(2h)检测RNA。
(3)磁珠吸附杂交:Gen– probe公司最近应用吖啶翁酯(acridinium ester)标记DNA探针,这种试剂可用更敏感的化学发光来检测。探针和靶杂交后,杂交物可特异地吸附在磁化的有孔小珠(阳离子磁化微球体上)。溶液中的磁性小珠可用磁铁吸出,经过简单的漂洗步骤,吸附探针的小珠可用化学发光测定。
2.发光液相杂交
(1)能量传递法:Heller等设计用两个紧接的探针,一个探针的一端用化学发光基团(供体)标记,另一个探针的一端用荧光物质标记,并且这两个探针靠得很近。两个靠得很近的探针用不同的物质标记(标记光发射),当探针与特异的靶杂交后,这些标记物靠得很近。一种标记物发射的光被另一种标记物吸收,并重新发出不同波长的光,调节 检测器使自动记录第二次发射光的波长。只有在两个探针分子靠得近时,才能产生受激发光,因此这种方法具有较好的特异性。
(2)吖啶翁酯标记法:吖啶翁酯标记探针与靶核酸杂交后,未杂交的标记探针分子上的吖啶翁酯可以用专门的方法选择性除去,所以杂交探针的化学发光是与靶核酸的量成比例的。该法的缺点是检测的敏感度低(约1ng的靶核酸),仅适用于检测扩增的靶序列,如rRNA或PCR扩增产物。
3.液相夹心杂交
(1)亲合杂交:在靶核酸存在下,两个探针与靶杂交,形成夹心结构,杂交完成后,杂交物可移到新的管或凹孔中,在其中杂交物上的吸附探针可结合到固相支持物上,而杂交物上的检测探针可产生检测信号。用生物素标记吸附探针,用125I标记检测探针,这个系统的敏感性可检测出4×106靶分子。该试验保持了固相夹心杂交的高度特异性。
(2)采用多组合成探针和化学发光检测:第一类探针是未标记的检测探针和液相吸附探针,它们有50个碱基长,其中含有30个细菌特异序列碱基和20个碱基的单链长尾;第二类探针是固相吸附探针,它可吸附在小珠或微孔板上。未标记检测探针的单链长尾用于结合扩增多个标记探针,液相吸附探针和靶杂交物从溶液中分离并固定在小珠或微板上,典型的试验可用25个不同的检测探针和10个不同的吸附探针。第一个标记检测探针上附着很多酶(碱性磷酸酶或过氧化物酶)可实现未标记检测探针的扩增。使用化学发光酶的底物比用显色反应酶的底物更敏感。这个杂交方法已用于乙肝病毒、沙眼衣原体、淋球菌以及质粒抗性的检测,敏感性达到能检测5×104双链DNA分子。
4.复性速率液相分子杂交这个方法的原理是细菌等原核生物的基因组DNA通常不包含重复顺序。它们在液相中复性(杂交)时,同源DNA比异源DNA的复性速度要快。同源程度越高,复性速率和杂交率越快。利用这个特点,可以通过分光光度计直接测定变性DNA在一定条件下的复性速率,进而用理论推导的数学公式来计算DNA-DNA之间的杂交(结合)度。
六、核酸分子杂交实验因素的优化
(一)探针的选择
根据不同的杂交实验要求,应选择不同的核酸探针。在大多数情况下,可以选择克隆的DNA或cDNA双链探针。但是在有些情况下,必须选用其它类型的探针如寡核苷酸探针和RNA探针。例如,在检测靶序列上的单个碱基改变时应选用寡核苷酸探针,在检测单链靶序列时应选用与其互补的DNA单链探针(通过克隆人M13噬菌体DNA获得)或RNA探针,寡核苷酸探针也可。长的双链DNA探针特异性较强,适宜检测复杂的靶核苷酸序列和病原体,但不适宜于组织原位杂交,因为它不易透过细胞膜进入胞内或核内。在这种情况下,寡核苷酸探针和短的PCR标记探针(80~150bp)具有较大的优越性。
在选用探针时经常会受到可利用探针种类的限制。如在建立DNA文库时,手头没有筛选特定基因的克隆探针,这时就可用寡核苷酸探针来代替。但必须首先纯化该基因的编码蛋白,并测定6个以上的末端氨基酸序列,通过反推的核苷酸序列合成一套寡核苷酸探针。如果已有其它动物的同种基因克隆,因为人类和动物间在同一基因的核苷酸顺序上存在较高的同源性,因此可利用已鉴定的动物基因作探针来筛选人类基因克隆。对于基因核苷酸序列背景清楚而无法获得克隆探针时,可采用PCR方法扩增某段基因序列,并克隆人合适的质粒载体中,即可得到自己的探针。这种方法十分简便,无论基因组DNA探针还是cDNA探针都可以容易地获得,而且,可以建立PCR的基因检测方法,与探针杂交方法可作对比,可谓一举两得。
(二)探针的标记方法
在选择探针类型的同时,还需要选择标记方法。探针的标记方法很多,选择什么标记方法主要视个人的习惯和可利用条件而定。但在选择标记方法时,还应考虑实验的要求,如灵敏度和显示方法等。一般认为放射性探针比非放射性探针的灵敏度高。放射性探针的实际灵敏度不依赖于所采用的标记方法,如随机引物延伸法往往得到比缺口平移法更高的比活性。在检测单拷贝基因序列时,应选用标记效率高、显示灵敏的探针标记方法。在对灵敏要求不高时,可采用保存时间长的生物素探针技术和比较稳定的碱性磷酸酶显示系统。
(三)探针的浓度
总的来说,随探针浓度增加,杂交率也增加。另外,在较窄的范围内,随探针浓度增加,敏感性增加。依我们的经验,要获得较满意的敏感性,膜杂交中32P标记探针与非放射性标记探针的用量分别为5~10ng/ml和25~1000ng/ml,而原位杂交中,无论应用何种标记探针,其用量均为0.5~5.0μg/ml。探针的任何内在物理特性均不影响其使用浓度,但受不同类型标记物的固相支持物的非特异结合特性的影响。
(四)杂交率
传统杂交率分析主要用于DNA复性研究,在这种情况下,探针和靶链在溶液中的浓度相同。现代杂交实验无论在液相杂交还是固相杂交均在探针过剩的条件下进行,此外,固相杂交中靶序列不在液相,故其浓度不能精确计算。因此,本文不讨论通常用于杂交反应的传统二级速率公式,而叙述一级动力学公式。
在探针过量的条件下,杂交率主要依赖于探针长度(复杂度)和探针浓度。下面列出的公式适用于过剩单链探针对靶序列杂交的情形,双链探针开始时(1~4h),杂交动力学相同,但长时间杂交后,由于探针本身的复性,可用于杂交的探针浓度会逐渐降低。公式(1)可用于估计半数探针与固定靶序列杂交所需的时间。
t1/2=ln2/kc
t=保温(杂交)时间(s);k =形成杂交体的速率常数[mol/(Lxntxs)];c =溶液中的探针浓度(mol/L)。速率常数K决定于探针长度(L)、探针复杂度(N)、温度、离子强度、粘度和pH。不含重复序列的探针,l =N。例如,对一个含两个20nt序列的40mer探针而言,l=40,N=20。K与这些变量的关系为:
Kn= 3.5×105K=KnL0.5/N (2)
Kn是缔结常数,Kn =3.5×105。Na+浓度为0.4~1.0mol/L, Ph5~9和杂交温度低于探针-靶序列杂交体Tm值2.5℃时,公式(1)和(2)可合并为(3),用于计算半数探针与靶序列的杂交率(以秒计)。
对一个长500个碱基的探针而言,此值为:
长500个碱基的探针杂交时间很长(20h),应用短探针和使用杂交促进剂有其优越性。由于实际应用的探针长度变化较大(对>1kb的探针,因扩散与粘度效应不可能使因素L得到合适的补偿)。另外,固靶序列也不可能都用于杂交,所以,由公式预计的随探针长度增加的杂交率不一定总是正确的。
(五)杂交最适温度
杂交技术最重要的因素之一是选择最适的杂交反应温度。若反应温度低于Tm 10~15℃,碱基顺序高度同源的互补链可形成稳定的双链,错配对减少。若反应温度再低(Tm-30℃),虽然互补链之间也可形成稳定的双链,但互补碱基配对减少,错配对增多、氢键结合的更弱。如两个同源性在50%左右或更低些的DNA,调整杂交温度可使它们之间的杂交率变化10倍,因此在实验前必须首先确定杂交温度。通常有三种温度可供试验,即最适复性温度、苛刻复性温度及非苛刻复性温度。温度的选择及温度对杂交的影响见表18-3。最适复性温度(Optimunm renaturationtemperature, TOR):Tor =Tm –25℃
苛刻复性温度:Ts = Tm – (10或15℃)
非苛刻复性温度:Tns =Tm – (30或35℃)
在2×SSC反应液中,可以根据下列公式计算最适复性温度:TOr =0.51 (G+C%)+47℃。
表18-4 DNA—DNA杂交温度的选择范围(2×SSC)
| DNA中G+C mol% | 杂交反应温度(℃) | ||
| TOR | Ts | Tns | |
| 30 | 62.3 | 73.3 | 52.3 |
| 35 | 64.9 | 74.9 | 54.9 |
| 40 | 67.4 | 77.4 | 57.4 |
| 45 | 70.0 | 80.0 | 60.0 |
| 50 | 72.5 | 82.5 | 62.5 |
| 55 | 75.1 | 85.1 | 65.1 |
| 60 | 77.6 | 87.6 | 67.6 |
| 65 | 80.2 | 90.2 | 70.2 |
| 70 | 82.7 | 92.2 | 72.7 |
| 75 | 85.3 | 95.3 | 75.3 |
可以看出DNA复性和DNa–RNA, DNA-DNA杂交通常要在高温反应条件下进行,其反应的最大速度是在低于Tm值约25℃。然而对于那些反应时间需要延长,或对生物活性必须保护的复杂生物的核酸研究(如哺乳动物),核酸长时间处于高温下很显然是不利的。这会引起核酸链的断裂、胶嘌呤的作用,结合到膜上的DNA脱落也会增多。这个问题可以通过使用高浓度盐溶液(如6.2mol/l NaCl),或使用某些有机溶剂的水溶液降低反应温度来解决。常使用的有机溶剂有两类,甲酰胺和二甲亚砜(DMSO)。在杂交液中加入30%二甲亚砜可使T2噬菌体DNA的Tm值比原先降低14℃,而使用酰胺甚至可使DNA在室温下变性和复性。Mc-Conaughy等发现,反应液中每增加1%的甲酰胺浓度,Tm值可降低0.72℃。
现在认为,适当选择甲酰胺和盐水浓度及合适的反应温度,可使DNA复性和DNA-RNA杂交获得高特异性和更快的反应速度。
(六)杂交的严格性
影响杂交体稳定性的因素决定着杂交条件的严格性。一般认为在低于杂交体Tm值25℃时杂交最佳,所以首先要根据公式(4)计算杂交体Tm 值。由此式可见,通过调节 盐浓度、甲酰胺浓度和杂交温度来控制所需的严格性。对用20个碱基以上的探针做DNA:DNA杂交的Tm值计算如下:
n =杂交体中最短链的长度,因此,对一个G+C为42%的500个碱基探针于5×SSC(0.75mol/l Na+)和50%甲酰胺杂交的Tm 值为:
T=81.5 +(-2.07)+ 17.22 –1 –(30.5)=65℃
T杂交 =65℃–25℃=40℃
影响TM值的其它因素:
(1)对克隆或合成探针而言,同源性每下降1%,Tm值就降低1.5℃,15~50个碱基的寡核苷酸探针的这种作用更明显。
(2)RNA:DNA杂交体的Tm值较同样的DNA:DNA杂交体的高10~15℃。
(3)RNA:RNA杂交体的Tm值较同样的DNA:DNA杂交体的高20~25℃。
显然,当用RNA为靶序列时,要使用甲酰胺来降低Tm 值以保证合适的杂交温度。当以克隆的探针进行膜杂交时,在最后的漂洗步骤中应达到最严格的条件。对一个500个碱基探针而言,典型最终漂洗条件点0.1×SSC(0.015mol/l Na+),55℃。代入公式(4)可得:
Tm =81.5 (-30.3) +17.22 –1-0 =67℃
67℃-55℃=12℃
因此,较Tm低12℃的漂洗条件比Tm 低25℃的杂交相比条件更严格了。
对寡核苷核探针而言,杂交温度往往低于Tm5℃,因此,对一个G+C为50%的30nt寡核苷酸探针来说,Tm 值为:
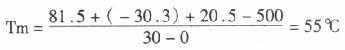
T杂交=55℃-5℃=50℃
下面一个经典的公式适用于14-20个碱基的寡核苷酸探针:
Tm =4℃(g + C) +2℃(a + T)
在实际应用中,寡核苷酸探针的最佳杂交温度必须精确确定。最方便的一种方法是制备一张含不同稀释度靶DNA和非特异靶DNA(如鱼精或大肠杆菌DNA)的膜。在不同温度下使膜与探针杂交,特异靶序列结合探针信号很强,而非特异靶序列与探针无任何反应的温度就是最适温度,在某些条件下,可用二甲亚砜(DMSO)代替甲酰胺来降低Tm值。
用一个以上的探针的(如夹心杂交)杂交系统中,估计Tm值更加复杂。可用上述公式估计每一探针的Tm 值。然后求其均值作为杂交温度。
(七)杂交反应时间
在条件都得到满足的情况下,杂交的成败就取决于保温时间。时间短了,杂交反应不完成;时间长了也无益,会引起非特异结合增多。一般杂交反应要进行20h左右。1966年Britten和Kohne推荐用Cot =值来计算杂交反应时间。Cot 值实际上是杂交液中单链起始浓度(Co)和反应时间(t)的乘积。实验表明Cot =100时,杂交反应基本完成。Cot=0,基本上没有 杂交。例如在液相杂交中未标记的DNa 400μg/ml(按单股DNA每微克 紫外吸收值为0.024计算,总的吸收值为9.6),如果反应时间为21h,那么对于未标记的DNA来说,Cot =9.6/21 =100.8, 杂交完成了。对标记Dn A(浓度为0.1μg/ml)来说Cot值为0.05,这就充分排除了标记DNA的自我复性。
(八)杂交促进剂
惰性多聚体可用来促进250个碱基以上的探针的杂交率。对单链探针可增加3倍,而对双链探针、随机剪切或随机引物标记的探针可增加高达100倍。而短探针不需用促进剂,因其复杂度低和分子量小,短探针本身的杂交率就高。
硫酸葡聚糖是一种广泛用于较长双链探针杂交的促进剂。这是一种多聚胺,平均分子量为500000。另一种常见的促进剂是聚乙二醇(PEG),PEG分子量小(6000~8000)、粘度低、价格低廉,但它不能完成取代硫酸葡聚糖。在某些条件下5%~10%硫酸葡聚糖效果较好,若用5%~10%PEG则可产生很高的本底。因此,使用促进剂时有必要优化条件。另一种多聚体促进剂是聚丙烯酸,用其钠盐,浓度为2%~4%。与硫酸葡聚糖相比,其优点是价格低廉,粘度低(MW=90000)。
小分子化学试剂酚和硫氰酸胍也能促进杂交,它们可能是通过增加水的疏水性和降低双链和单链DNA间的能量差异而发挥作用。酚作为杂交促进剂,只能在低DNA浓度的液相杂交中观察到,该方法曾被称为酚乳化复性技术,该法不能用于固相杂交,因酚可引起核酸与膜的非特异吸附作用,即使在液相杂交中的应用也是有限的。而硫氰酸胍可通过降低双链DNA的Tm值而起作用。此外,该分子还可以促进RNA的杂交,有裂解细胞而抑制RNase的作用。总之,硫酸葡聚糖和聚乙二醇因能用于固相杂交是目前最常用的杂交促进剂。
第四节 石蜡包埋组织的DNA提取及其应用
近10年来,现代分子生物学技术越来越广泛地被用于人类疾病研究的诸领域,为了解病理状态下基因组DNA的变化积累了新资料。目前认为,人类基因组并非人们想象的那样稳定,诸如基因重排、扩增、缺失,突变和DNA甲基化类型改变等时有发生,这些改变对于基因表达和调控,以及疾病过程的发展与转归等方面均具有重要意义。
医院病理科档案中积存的大量石蜡包埋组织,是一个可靠的分子生物学研究的材料来源。在石蜡切片上进行原位杂交或原位PCR分析的分子生物学方法,是免疫细胞化学技术的重要延伸。通过对DNA或mRNA分析亦可直接证实或补充免疫细胞化学的发现。这些对形态学家较为熟悉的原位分子生物学技术,本节 不再赘述。Goelz(1985)和Dubeau 等(1986)成功地从蜡块中提取出高质量的DNA,完全可以满足某些肿瘤分子生物学研究的需要,结束了DNA研究依赖于新鲜或冰冻组织和细胞的历史,而且可以广泛地应用于大宗病例的回顾性研究,对肿瘤发生的分子机制的探讨,诊断与鉴别诊断研究和患者预后评估等方面均具有重要价值。本节 重点讨论石蜡包埋组织DNA提取的方法学及其潜在的应用前景。
一、石蜡包埋组织DNA提取的基本方法
从石蜡包埋组织中提取DNA的基本步骤,是在常规DNA提取方法的基础上改良和演变而来。Goelz等(1985)最先提出的石蜡包埋组织DNA提取技术(表18-5),是用机械的方法破碎组织,切除多余的石蜡,直接进入含SDS和高浓度蛋白酶K(1mg/ml)的提取缓冲液加温孵育,然后进行苯酚和氯仿抽提。所得DNA虽不完整,但可被限制性内切酶切割,因而适于点杂交,印迹杂交等多种分子生物学实验。Dubeau等(1986)在上述方法上作了改进。首先通过组织切片用二甲苯脱蜡,保证了组织的完全破碎,使细胞与消化液充分接触,使DNA释放和蛋白去除更加完全;其次通过两步消化的方法,将不适于做分子杂交的降解的DNA小片段去除,仅保留那些可螺旋化的完整的DNA大分子,从而提高了DNA质量,适于做Southern印迹杂交分析。Moerkerk等(1990)对上述两种方法进行了比较,发现两种方法所取得DNA之点杂交结果一致,非螺旋化DNA亦不影响杂交分析。
表18-5 Gpelz氏DNA提取方法的主要步骤
| 1.切除多余石蜡,暴露组织 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
2.将组织切碎(最大径109);而若X=80%时,则y=1.830=45517159.6(>107)。由此可见,其扩增的倍数是巨大的,将扩增产物进行电泳,经溴化乙锭染色,在紫外灶照射下(254nm)一般都可见到DNA的特异扩增区带。第三节 PCR操作范例及反应体系的组成一、PCR操作范例在一个典型的PCR反应体系中需加入:适宜的缓冲液、微量的模板DNA、4×dNTPs、耐热性多聚酶、Mg2+和两个合成的DNA引物。模板DNa 94℃变性1min,引物与模板40~60℃退火1min,72℃延伸2min。在首次循环前模板预变性3~5min;在末次循环后,样品仍需继续延伸3~5min以上,确保扩增的DNA为双链DNA。为便于了解PCR反应中各成份的组成,加入量和反应条件,使人们以此为基础,对不同的研究对象逐项改变来找到最佳反应条件,特列举Perkin Elmer Cetus公司Gene Amp DNA试剂盒提供的典型反应条件供参考。 表22-1 PCR反应混合液
扩增条件:94℃60s,37℃60s,72℃120s,共25~30个循环。 注:[1]反应缓冲液[10×]含:100mmol/l Tris-HCl pH8.3(25℃), 15mmol/L KCl, 15mmol/L MgCl2, 0.01%(W/V)明胶(Sigma G2500) [2]酶储存缓冲液(-20℃)含:50%甘油,100mmol/l KCl, 20mmol/L Tris-HCl ph8.0 0.1mmol/L EDTA, 1.0mmol/L DTT 200μg/ml明胶 0.5%吐温20,0.5% Nonidet P40 [3][4]引物,1,2:扩增λ-噬菌体基因中500bp的片段 引物1序列:7131~7155(5’)-GATGAGTTCGTGTCCGTACAACTGG-(3’) 引物2序列:7606~7630(5’)-GGTTATCGAAATCAGCCACAGCGCC-(3’) 注意(3’)端有2个bp互补故易生成50bp的双体 二、PCR反应系统的组成(一)PCR缓冲液(PCrBuffer)用于PCR的标准缓冲液见PCR操作范例。于72℃时,反应体系的pH值将下降1个单位,接近于7.2。二价阳离子的存在至关重要,影响PCR的特异性和产量。实验表明,Mg2+优于Mn2+,而Ca2+无任何作用。 1.Mg2+浓度Mg2+的最佳浓度为1.5mmol/L(当各种dNTP浓度为200mmol/L时),但并非对任何一种模板与引物的结合都是最佳的。首次使用靶序列和引物结合时,都要把Mg2+浓度调到最佳,其浓度变化范围为1~10mmol/L。Mg2+过量易生成非特异性扩增产物,Mg2+不足易使产量降低。 样品中存在的较高浓度的螯合剂如EDTA或高浓度带负电荷的离子基团如磷酸根,会与Mg2+结合而降低Mg2+有效浓度。因此,用作模板的DNA应溶于10mmol/l Tris-HCl(pH7.6)0.1mmol/L EDTA中。 dNTP含有磷酸根,其浓度变化将影响Mg2+的有效浓度。标准反应体系中4×dTNPs的总浓度为0.8mmol/L,低于1.5mmol/L的Mg2+浓度。因此,在高浓度DNA及dNTP条件时,必须相应调整Mg2+的浓度。 2.Tris -HCl缓冲液在PCR中使用10~50mmol/L的Tris –HCl缓冲液,很少使用其他类型的缓冲液。Tris缓冲液是一种双极化的离子缓冲液,pKa为8.3(20℃),△pKa为0.021/℃。因此,20mmol/l Tris pH8.3(20℃)时,在典型的热循环条件下,真正的pH值在7.8~6.8之间。 3.KCl浓度K+浓度在50mmol/L 时能促进引物退火。但现在的研究表明,NaCl浓度在50mmol/L时,KCl浓度高于50mmol/L将会抑制Taq酶的活性,少加或不加KCl对PCR结果没有太大影响。 4.明胶明胶和BSA或非离子型去垢剂具有稳定酶的作用。一般用量为100μg/ml,但现在的研究表明,加或不加都能得到良好和PCR结果,影响不大。 5.二甲基亚砜(DMSO)在使用Klenow片段进行PCR时DMSO是有用的;加入10%DM-SO有利于减少DNA的二级结构,使(G+C)%含量高的模板易于完全变性,在反应体系中加入DMSO使PCR产物直接测序更易进行,但超过10%时会抑制Taq DNA聚合酶的活性,因此,大多数并不使用DMSO。 (二)四种脱氧三磷酸核苷酸(4×dNTPs)在PCR反体系中dNTP终浓度高于50mmol/L会抑制Taq酶的活性,使用低浓度dNTP可以减少在非靶位置启动和延伸时核苷酸错误掺入,高浓度dNTPs易产生错误掺入,而浓度太低,势必降低反应物的产量。PCR常用的浓度为50~200μmol/L,不能低于10~15μmol/L。四种dNTP的浓度应相同,其中任何一种浓度偏高或偏低,都会诱导聚合酶的错误掺入,降低合成速度,过早终止反应。 决定最低dNTP浓度的因素是靶序列DNA的长度和组成,例如,在100μl反应体系中,4×dNTPs浓度若用20μmol/L,基本满足合成2.6μg DNA或10pmol的400bp序列。50μmol/L的4×dNTPs可以合成6.6μgDNA,而200μmol/L足以合成25μg/DNA。 购自厂商的dNTP溶液一般均未调pH,应用1mol/l NaOH将dNTP贮存液pH调至7.0,以保证反应的pH值不低于7.1。市购的游离核苷酸冻干粉,溶解后要用NaOH中和,再用紫外分光光度计定量。 (三)引物的量引物在PCR反应中的浓度一般在0.1~1μmol/L之间。浓度过高易形成引物二聚体且产生非特异性产物。一般来说用低浓度引物经济、特异,但浓度过低,不足以完成30个循环的扩增反应,则会降低PCR的产率。 (四)TaqDNA聚合酶的量典型PCR反应混合物中,所用酶浓度为2.5U/μl,常用范围为1~4U/100μl。由于DNA模板的不同和引物不同,以及其它条件的差异,多聚酶的用量亦有差异,酶量过多会导致非特异产物的增加。 由于生产厂家所用兵配方、制造条件以及活性定义不同,不同厂商供应的TaqDNA聚合酶性能也有所不同。 Cetus公司酶定义是:1个酶单位是指在以下分析条件下,于74℃,30min内使10nmmol的dNTP掺入酸不溶性成分所需的酶。测定时间为10min,折算成30min掺入量。 分析条件为25nmol/L TAPS(三羟基-甲基-氨基丙烷磺酸钠pH9.3,25℃),50mmol/l KCl, 2mmol/L MgCl2,1mmol/L β-ME(巯基乙醇),dATP、dTTP、dGTP各200mmol/L,dCTP为100mmol/L(由不标记及α-32P标记混合),12.μg变性鲱鱼精子DNA,最终体积50μl。 (五)模板单、双链DNA或RNA都可以作为PCR的样品。若起始材料是RNA,须先通过逆转录得到第一条cDNA。虽然PCR可以仅用极微量的样品,甚至是来自单一细胞的DNA,但为了保证反应的特异性,还应用ng级的克隆DNA,μg水平的单拷贝染色体DNA或104拷贝的待扩增片段作为起始材料,模板可以是粗品,但不能混有任何蛋白酶、核酸酶、Taq DNA聚合酶抑制剂以及能结合DNA的蛋白。 DNA的大小并不是关键的因素,但当使用极高分子量的DNA(如基因组的DNA时),如用超声处理或用切点罕见的限制酶(如Sal1和Not1)先行消化,则扩增效果更好。闭环靶序列DNA的扩增效率略低于线状DNA,因此,用质粒作反应模板时最好先将其线状化。 模板靶序列的浓度因情况而异,往往非实验人员所控制,实验可按已知靶序列量逆减的方式(1ng,0.1ng,0.001ng等),设置一组对照反应,以检测扩增反应的灵敏度是否符合要求。 (六)石蜡油PCR扩增时建议在混合物上面铺一层石蜡油,减少PCR过程中尤其是变性时液体蒸发所造成的产物的丢失。研究表明,应用石蜡油可使扩增产量增加5倍,可能与石蜡油维持热恒定和整个反应体系中盐浓度有关。 三、电泳分析在实际工作中常采用琼脂糖凝胶电泳。一般情况下先在电泳缓冲液或凝胶中加1%溴化乙锭(EB)(每100ml加100μl),然后将已经制备好的1%~2%琼脂糖凝胶(用电泳缓冲液配制)放入电泳槽内,加入待测样品10μl,同时用分子量标准品作标记。琼脂糖浓度应按分离DNA片段的大小进行选择,一般用1.5%~2%,电泳电压75V,待样品进行凝胶内距胶末端1cm时,切断电源,取出凝胶在紫外灯下直接观察结果。 由于溴化乙锭可与双链DNA形成结合物,在紫外灯下能发射荧光,使EB的荧光强度增强80~100倍,所以,电泳后凝胶在紫外灯下可直接观察。一般肉眼观察DNA量可达10ng,其荧光强度与DNA含量成正比。 DNA分子在凝胶中泳动速度决定于电荷效应及分子效应。前者由所带净电荷量决定,而后者与分子大小及构型有关。按照DNA分子大小,其凝胶浓度可做不同的调整。有条件的实验室也可用聚丙烯酰胺凝胶电泳(PAGE)分析扩增的DNA片段。 表22-2 电泳检测扩增结果,EB荧光显色(254nm)
第四节 影响PCR的主要因素 PCR技术必须有人工合成的合理引物和提取的样品DNA,然后才进行自动热循环,最后进行产物鉴定与分析。引物设计与合成目前只能在少数技术力量较强的研究院、所进行,临床应用只需购买PCR检测试剂盒就可开展工作,PCR自动热循环中影响因素很多,对不同的DNA样品,PCR反应中各种成份加入量和温度循环参数均不一致。现将几种主要影响因素介绍如下。 一、温度循环参数在PCR自动热循环中,最关键的因素是变性与退火的温度。如操作范例所示,其变性、退火、延伸的条件是:94℃60s, 37℃60s, 72℃120s,共25~30个循环,扩增片段500bp。在这里,每一步的时间应从反应混合液达到所要求的温度后开始计算。在自动热循环仪内由混合液原温度变至所要求温度的时间需要30~60s,这一迟滞时间的长短取决于几个因素,包括反应管类型、壁厚、反应混合液体积、热源(水浴或加热块)以及两步骤间的温度差,在设置热循环时应充分给以重视和考虑,对每一仪器均应进行实测。 关于热循环时间的另一个重要考虑是两条引物之间的距离;距离越远,合成靶序列全长所需的时间也越长,前文给出的反应时间是按最适于合成长度500bp的靶序列拟定的。下面就各种温度的选择作一介绍。 1.模板变性温度变性温度是决定PCR反应中双链DNA解链的温度,达不到变性温度就不会产生单链DNA模板,PCR也就不会启动。变性温度低则变性不完全,DNA双链会很快复性,因而减少产量。一般取90~95℃。样品一旦到达此温度宜迅速冷却到退火温度。DNA变性只需要几秒种,时间过久没有必要;反之,在高温时间应尽量缩短,以保持Taq DNA聚合酶的活力,加入Taq DNA聚合酶后最高变性温度不宜超过95℃。 2.引物退火温度退火温度决定PCR特异性与产量;温度高特异性强,但过高则引物不能与模板牢固结合,DNA扩增效率下降;温度低产量高,但过低可造成引物与模板错配,非特异性产物增加。一般先由37℃反应条件开始,设置一系列对照反应,以确定某一特定反应的最适退火温度。也可根据引物的(G+C)%含量进行推测,把握试验的起始点,一般试验中退火温度Ta(annealing temperature)比扩增引物的融解温度TTm(meltingtemperature)低5℃,可按公式进行计算: Ta = Tm -5℃= 4(G+C)+ 2(A+T)-5℃ 其中A,T,G,C分别表示相应碱基的个数。例如,20个碱基的引物,如果(G+C)%含量为50%时,则Ta的起点可设在55℃。在典型的引物浓度时(如0.2μmol/L),退火反应数秒即可完成,长时间退火没有必要。 3.引物延伸温度温度的选择取决于Taq DNA聚合酶的最适温度。一般取70~75℃,在72℃时酶催化核苷酸的标准速率可达35~100个核苷酸/秒。每分钟可延伸1kb的长度,其速度取决于缓冲溶液的组成、pH值、盐浓度与DNA模板的性质。扩增片段如短于150bp,则可省略延伸这一步,而成为双温循环,因Taq DNA聚合酶在退火温度下足以完成短序列的合成。对于100~300bp之间的短序列片段,采用快速、简便的双温循环是行之有效的。此时,引物延伸温度与退火温度相同。对于1kb以上的DNA片段,可根据片段长度将延伸时间控制在1~7min,与此同时,在PCR缓冲液中需加入明胶或BSA试剂,使Taq DNA聚合酶在长时间内保持良好的活性与稳定性;15%~20%的甘油有助于扩增2.5kb左右或较长DNA片段。 4.循环次数常规PCR一般为25~40个周期。一般的错误是循环次数过多,非特异性背景严重,复杂度增加。当然循环反应的次数太少,则产率偏低。所以,在保证产物得率前提下,应尽量减少循环次数。 扩增结束后,样品冷却并置4℃保存。 二、引物引物设计要扩增模板DNA,首先要设计两条寡核苷酸引物,所谓引物,实际上就是两段与待扩增靶DNA序列互补的寡核苷酸片段,两引物间距离决定扩增片段的长度,两引物的5’端决定扩增产物的两个5’末端位置。由此可见,引物是决定PCR扩增片段长度、位置和结果的关键,引物设计也就更为重要。 引物设计的必要条件是与引物互补的靶DNA序列必须是已知的,两引物之间的序列未必清楚,这两段已知序列一般为15~20个碱基,可以用DNA合成仪合成与其对应互补的二条引物,除此之外,引物设计一般遵循的原则包括: 1.引物长度根据统计学计算,长约17个碱基的寡核苷酸序列在人的基因组中可能出现的机率的为1次。因此,引物长度一般最低不少于16个核苷酸,而最高不超过30个核苷酸,最佳长度为20~24个核苷酸。这样短的寡核苷酸在聚合反应温度(通过72℃)下不会形成稳定的杂合体。有时可在5’端添加不与模板互补的序列,如限制性酶切位点或启动因子等,以完成基因克隆和其他特殊需要;引物5’端生物素标记或荧光标记可用于微生物检测等各种目的。 有时引物不起作用,理由不明,可移动位置来解决。 2.(G+C)%含量引物的组成应均匀,尽量避免含有相同的碱基多聚体。两个引物中(G+C)%含量应尽量相似,在已知扩增片段(G+C)%含量时宜接近于待扩增片段,一般以40%~60%为佳。 3.引物内部应避免内部形成明显的次级结构,尤其是发夹结构(hairpinstructures)。例如:
4.引物之间两个引物之间不应发生互补,特别是在引物3’端,即使无法避免,其3’端互补碱基也不应大于2个碱基,否则易生成“引物二聚体”或“引物二倍体”(Primer dimer)。所谓引物二聚体实质上是在DNA聚合酶作用下,一条引物在另一条引物序列上进行延伸所形成的与二条引物长度相近的双链DNA片段,是PCR常见的副产品,有时甚至成为主要产物。 另外,两条引物之间避免有同源序列,尤为连续6个以上相同碱基的寡核苷酸片段,否则两条引物会相互竞争模板的同一位点;同样,引物与待扩增靶DNA或样品DNA的其它序列也不能存在6个以上碱基的同源序列。否则,引物就会与其它位点结合,使特异扩增减少,非特异扩增增加。 5.引物3’端配对DNA聚合酶是在引物3’端添加单核苷酸,所以,引物3’端5~6个碱基与靶DNA的配对要求必须精确和严格,这样才能保证PCR有效扩增。 引物设计是否合理可用PCRDESN软件和美国PRIMER软件进行计算机检索来核定。 人工合成的寡核苷酸引于最好经过色谱(层析)纯化或PAGE纯化,以除去未能合成至全长的短链等杂质。纯化引物在25%乙腈溶液中4℃保存可阻止微生物的生长;一般情况下,不用的引物应保存在-20℃冰箱中,在液体中引物能保存6个月,冻干后可保存1~2年。 三、DNA聚合酶早在1956年Kornberg等就从大肠杆菌提取液中发现了DNA聚合酶,并且得到了DNA聚合酶Ⅰ纯品。DNA聚合酶Ⅰ是由分子量为109000的一条多肽链构成,此酶可被枯草杆菌蛋白酶分解为两个片段,一个片段分子量为76000,有聚合酶活性,并有3’→5外切酶活力,即Klenow片段(Klenow fragment)。另一个片段分子量为34000,具有5’→’3’外切酶活力。因此,DNA聚合酶具有几种功能:一是聚合作用,以DNA为模板,将dNTP中的脱氧单核苷酸逐个加到3-OH末端。二是有’3’→5’外切酶活力,能识别和消除错配的引物末端,与复制过程中校正功能有关。三是5’→3’外切酶活力,它能从5’端水解核苷酸,还能经过几个核苷酸起作用,切除错配的核苷酸。1985年Mullis 等发明了PCR方法,以Klenow片段完成β-珠蛋白的PCR后,世界上许多实验室就考虑用耐热DNA聚合酶代替Klenow片段进行PCR,使耐热多聚酶的研究得以迅速发展。人们从生活于60℃(B.Stearothermophilus)到87℃(S.Solfatavicus)的许多菌中分离纯化出耐热DNA聚合酶,但有些酶不能耐受DNA变性所需温度,所以无法应用于PCR。现就PCR反应中常用的DNA聚合酶等作一详细介绍。 1.Taq DNA聚合酶用Taq DNA聚合酶代替大肠杆菌DNA聚合酶Ⅰ的Klenow片段是使PCR普及应用的关键。Klenow片段不能耐受95℃的双链DNA变性温度,所以每次循环都要加入新酶;而Taq DNA聚合酶可以耐受93~95℃的高温,避免了不断补加多聚酶的繁琐操作,同时使退火和延伸温度得以提高,减少了非特异性产物和DNA二级结构对PCR的干扰,增进了PCR特异性、产量和敏感度,二者相比,其主要区别在于:①Klenow酶的最适温度为37℃,扩增的产物并非全是目的序列,需用探针检测。Taq酶则不仅产率高而特异性也高。它的最适温度为74~75℃。因而使退火温度可以提高,使退火严格性提高,减少错配引物的延伸。②循环后期酶量渐感不足而产生平坡。到达平玻的循环次数,Klenow酶为20个(均用1μg基因组DNA开始)而Taq酶为30个。③延伸片段长度Taq酶为10kb以内,而Klenow酶为400bp以内。 Taq酶由水栖高温菌(Thermusaquatics)YT1蓖株中分离而得。此菌于1969年由Brock分离自美国黄石公园温泉,作为栖热杆菌的标准菌株,其生长温度为70~75℃。最初从中分离到分子量60~68KDa,比活性为2000~8000U/mg的DNA聚合酶。后来Cetus公司的Kary Mullis等又分离到比活为20万U/mg的纯酶,分子量为93910。此种9.4KDa酶的最适温度为75~80℃,与单纯核苷酸的结合率(Kcat)可达150核苷酸(nt)/s酶分子。以M13模板,用富含G+C的30bp引物延伸,70℃时Kact>60nt/s;55℃可达24nt/s;37℃时为1.5nt/s,而22℃时低至0.25nt/s。高于90℃时DNA合成活性甚差,这种高温条件下,引物与模板已不能牢固结合。 在PCR反应混合液中,Taq酶于92.5℃,95℃及97.5℃保持其50%活力的时间分别为130、40及5~6min,在50次循环的PCR中当管内最高温度为95℃。每循环为20s时尚可保持65%活力。Taq 酶在95℃的半寿期为40min,故在PCR循环中选用的变性温度,不宜高于95℃。 Taq酶现已可用基因重组的方法生产,商品名为Ampli Taq(Cetus公司)。Taq酶的完整基因长2499bp,在大肠杆菌中表达生产,含832个氨基酸。在氨基酸序列上与大肠杆菌DNA聚合酶Ⅰ有38%是一致的,包括对dNTP结合,引物与模板作用区均存在于Taq酶中。 Taq酶具有依赖DNA合成的5’→’3’外切酶活性,因此,模板上有一段退火的3’-磷酸化的“阻断物”,会被逐个切除而不会阻止来自上游引物链的延伸,而对于5’-32P标记的合成寡核苷酸引物,则无论是单链或是与模板复性,都未发现降解,所以该种活性不会影响PCR结果。Taq酶没有3’→’5’外切酶活性,如果发生dNTP错误掺入,这种酶没有校正能力,因此运用Taq酶进行PCR,产物中点突变较多,对克隆等不太有利。一般错掺率为1.25×10-4~1×10-5(4×dNTPs浓度分别为200μmol/L,Mg2+为1.5mmol/L,在55℃退火)。但不含3’→5’外切酶活性对测序有利。 2.影响酶活力的因素Taq酶的活力受Mg2+离子的影响。用鲱精DNA为模板,总dNTP浓度0.7~0.8mmol/L,Mg2+为2.0mmol/L时激活能力最高。浓度超过此值产生抑制。10mmol/l MgCl2抑制活力达40%~50%。dNTP能与Mg2+结合,故游离Mg2+只是结合后剩余的量。若总dNTP浓度高至4~6mmol/L时,Taq酶活力要降低20~30%,即底物抑制。 dNTP浓度低时PCR产率及特异性均增高,适合于用扩增掺入法标记生物素及放射性元素。当100μl PCR液中含dNTP各40μmol/L时就足以合成2.6μg的DNA(dNTP消耗一半)。 表22-3 有机溶剂对Taq聚合酶活力的影响
|