第一章 概论
一、医学遗传学研究的对象和范围
医学遗传学(medical genetics)是医学与遗传学相结合的一门边缘学科,是遗传学知识在医学领域中的应用。而医学遗传学的理论和实践又丰富和发展了遗传学。医学遗传学的研究对象是人类。人类遗传学(human genetics)探讨人类正常性状与病理性状(trait,或character特征)的遗传现象及其物质基础。而医学遗传学则主要研究人类(包括个体和群体)病理性状的遗传规律及其物质基础。医学遗传学通过研究人类疾病的发生发展与遗传因素的关系,提供诊断、预防和治疗遗传病和与遗传有关疾病的科学根据及手段,从而对改善人类健康素质作出贡献。
医学遗传学不仅与生物学、生物化学、微生物及免疫学、病理学、药理学、组织胚胎学、卫生学等基础医学密切有关,而且已经渗入各临床学科之中。研究临床各种遗传病的诊断、产前诊断、预防、遗传咨询和治疗的学科称为临床遗传学(clinical genetics)。
医学遗传学主要由人类细胞遗传学(human cytogenetics)和人类生化遗传学(human biochemical genetics)组成。它们分别用形态学和生物化学方法研究人类正常及变异性状的物质基础。而分子遗传学(molecular genetics)是生化遗传学的发展和继续;分子细胞遗传学(molecular cytogenetics)则是细胞遗传学与分子遗传学结合的产物。它们互相补充,甚至正融为一体,使人们能从基因水平提示各种遗传病的本质,从而不断完善基因诊断、预防以至治疗遗传病的措施。与医学遗传学关系密切的其他遗传学分支还有:
群体遗传学(population genetics)研究群体中基因的行为。探讨人类正常和病理性状在群体中分布及变迁的规律,研究从群体水平对遗传病的防治作远期效果估价。群体细胞遗传学(population cytogenetics)和遗传流行病学(geneticepidemiology)是这一学科的分支
免疫遗传学(immunogenetics)研究免疫现象的遗传基础。从分子水平阐明人类免疫现象的遗传和变异规律以及与遗传有关免疫性疾病的遗传背景,以揭示生物免疫现象的本质及遗传控制。
药物遗传学(pharmacogenetics)是生化遗传学的一个分支。研究药物反应个体差异的遗传基础。在理论上,它从一个侧面阐明遗传易感性的物质基础;在实践上,为指导医生用药的个体化原则提供理论根据。
辐射遗传学(radiation genetics)研究辐射对生物产生遗传效应的规律。从进化来看,确定辐射对地球上所有生物的遗传效应,可以达到影响整个生物圈的进化过程。
毒理遗传学(toxico genetics)或称遗传毒理学(genetic toxicology)是用遗传学方法研究环境因素对遗传物质的损害、产生机制及子代影响的一门学科。具体包括致变(mutagenesis)、致癌(carcinogenesis)及致畸(teratogenesis)的“三致”效应及其检测和评价这类效应的一套手段。
体细胞遗传学(somatic cell genetics)通过体细胞,特别是离体培养的体细胞研究基因的作用。它对基因定位的调节、细胞分化、个体发育、肿瘤的发生以及基因治疗都提供了重要的研究手段。
行为遗传学(genetics of behavior)研究基因对人类和动物行为的影响。这门学科对阐明人类正常及异常的社会行为、个性、智力、神经病和精神病的发生和表现都极为重要。
发育遗传学(developmental genetics)研究基因对发育过程的控制与调节,研究基因在发育不同阶段的表达及调控机制。
肿瘤遗传学(cancer genetics)研究肿瘤发生发展的遗传因素,研究恶性变、发展、转移的遗传基础。它不仅有助于探讨肿瘤的病因和发病机制,而且对肿瘤的早期诊断、预后和防治提供科学根据。
基因工程(genetic engineering)基因工程是一种新技术,即将基因加以人工改造而表达为新性状的科学,在人类遗传病的基因诊断及基因治疗中有重要作用。
优生学(eugenics)是研究用遗传学的原理和手段来提高人类素质的一门科学。
二、医学遗传学的发展史
医学遗传学借助于现代生物学的研究方法,在遗传学理论指导和实验方法广泛采用的基础上发展起来的。人类在遗传学中获得的每一新的成就都非常迅速地应用于研究人类的疾病,因而医学遗传学近年来得以突飞猛进。
医学遗传学早期受孟德尔、摩尔根经典遗传学的指引,对遗传病的来源及传递方式作了朴实的描述。本世纪初,随着染色体制备技术和观察方法的建立,生物化学理论和实验手段的发展,人类细胞遗传学和生化遗传学才迅速成长。
1923-1952年,由于低渗制片技术的建立(徐道觉等)和使用秋水仙碱获得了更多中期细胞分裂象(蒋有兴等)后,才证实人体细胞染色数目为46。1959年相继发现先天愚型为21三体(Lejeune等)、Klinefelter综合征为47,XXY(Jacob和Strong)、Turner综合征为45,X等染色体改变,标帜着临床遗传学的建立。1970年Caspersson应用喹咔因氮芥荧光染色使每对染色体显示特殊带型(显带技术)。继后,Yunis(1978)应用同步培养法,使细胞分裂停留于中期之前各期,显示出更多带型(高分辨显带技术)。这样,对染色体序号的确认、对染色体上微细变化以致对染色体疾病的认识都不断深化。染色体脆性部位与脆性X综合征的研究开辟了细胞遗传学的新领域。荧光原位杂交(FISH)使细胞遗传学获得了新的应用方向。通过细胞遗传学与分子遗传学的结合,现在已能用显微切割(micro-dessection)的方法,切下染色体特定区带进行微克隆,进而认识某区带所含DNA顺序的结构和功能,这将有助于对遗传病特别是染色体病发生奥秘的认识。
人类生化遗传学的发展应追溯到1902年Garrod对尿黑酸尿症等病的观察。他认为某一代谢环节出现先天性差错(inborn error of metebolism)可以导致遗传病。1948年发出了黄递酶缺乏以及1952年证实糖原贮积病I型是由于葡萄糖-6-磷酸酶缺乏引起后才确认Garrod判断是正确的。这类疾病现称为遗传性酶缺陷或遗传性酶病(enzymopa-thy),目前已证实的有200多种。另一方面,Pauling(1949)在研究镰形细胞性贫血时发现电泳慢速的HbS,提出蛋白质分子的遗传变异可引致一类疾病,他称之为分子病(molecular disease)。1956年他的同事Ingram证实HbS是由于球蛋白β链单个氨基酸置换(β6谷→缬)引起。现知免疫球蛋白、胶原蛋白、膜蛋白、凝血因子等遗传变异均可产生分子病。目前已证实的分子病约有200种。70年代崛起的分子遗传学将遗传病的研究推向了一个新的阶段。一大批遗传病因都从分子水平得以阐明,并迅速对基因定位、基因诊断及产前诊断以至基因治疗取得丰硕成果。展望未来,现今医学遗传学正在研究的热门课题是:
1.人类基因组计划(human genomeproject)1986年Berg提出将人类基因组的核苷酸顺序全部“弄清”。在“弄清”的基础上还要“弄懂”这些DNA顺序代表什么意义。这是一个生命科学的“登月计划”,是“人类细胞的分子解剖学和生理学”。人类基因组据估计约有3×109碱基对。美国计划用35亿美元,现已联合日本、欧洲国家成立了人类基因组组织(Human Genome Organization,HUGO)来完成这一巨大工程。由于基因工程技术的进步(如大片段DNA的切割与分离,YAC重叠克隆系的建立,测序技术的快速化等)已在第二个五年计划的伊始就完成了70%-75%基因组建立YAC库的工作,Y及21号染色体的测序已大体完成,X染色体DNA顺序也即将弄清,发展比预期快。同时,“弄懂”的工作也已展开,基因定位的进展、位置克隆(positional cloning)、外显子捕获(exon trapping)等技术的建立大大加快了对编码蛋白基因的认识。这项工程的实施无疑将大大推动医学和人类遗传学的发展。(详见第七章)
2.基因定位 基因定位就是要将结构基因和有价值的DNA片段定位于染色体的某一区带,由此绘制出人类基因定位图。这对克隆新的基因、了解基因功能与调节、基因间相互关系、基因与进化以及阐明遗传病遗传方式、病因及发病机制、遗传咨询及产前诊断都极重要。据1993年人类基因制图(human gene mapping)第12次国际会议(HGM93)报道,已定位人类基因4000多个。(详见第七章)
3.遗传病病因及发病机制的阐明 尽管随着分子遗传学的发展,许多单基因遗传病的病因得到阐明,甚至发现其异质性。但目前发现的6000多种单基因病和性状中,从蛋白质或酶水平证实病因者不到1/10。这些疾病发病机制的研究仍是薄弱环节。对多基因病如动脉粥样硬化、精神分裂症、糖尿病等的分子水平研究仍在起始阶段。这些领域尚需加强。
4.肿瘤遗传学 肿瘤是危害人类健康的大患。近年来对癌基因(oncogene)、肿瘤抑制基因(tumor suppressor gene)以及肿瘤转移基因(metastatic gene)和肿瘤转移抑制基因(non-metastatic gene)的发现及深入研究无疑是对肿瘤的发生、恶性转化、转移的重大突破。但要彻底了解各种肿瘤的发生发展机制仍有很大距离。而这些基因研究将为肿瘤的防治奠定基础。(详见第九章)。
5.基因诊断 基因诊断,特别是基因产前诊断是目前预防遗传病的主要手段。日新月异的各种方法使基因诊断日臻完善和简化,目前正拓宽可用此法诊断遗传病的领域。原则上所有的单基因病都可能进行基因诊断,要达到此目标尚需做大量的工作。早期(植入前)和母血产前基因诊断成了现今的热门话题。(详见第十三章)。
6.基因治疗 基因治疗的目标是要用正常基因取代致病基因,达到根治遗传病的目的。目前这一工作已在许多实验室进行,并已取得瞩目的效果。有些遗传病已开始进入人体试验阶段,可望在本世纪末或下世纪初在个别病种取得突破。目前似乎人们更热衷于肿瘤的基因治疗(详见第十四章)。
我国医学遗传学的实验研究工作开始于60年代。1962年项维、吴旻等首先报告了中国人的染色体组型,标志着我国人类细胞遗传学的开始。在生化遗传学方面,当时已对血红蛋白病和红细胞葡萄糖6-磷酸脱氢酶(G6PD)缺乏症开展了实验性研究,标志着我国生化遗传学的萌芽。此后相当长一段时间,我国医学遗传学停滞不前。直到1979年底我国召开了第一次人类和医学遗传学论文报告会后,医学遗传学才得到迅猛发展,部分医学院校已将医学遗传学列入必修课或选修课,各地开办了各种形式的临床医生培训班。在原来的工作基础上又开展了先天性代谢缺陷、免疫遗传学、肿瘤遗传学、眼遗传病、神经精神遗传病、酶和蛋白质多态性、群体遗传学、遗传咨询以及诱变剂检测等工作。80年代后期,我国处于前沿的细胞遗传学,引进了先进的高分辨显带技术、显微切割及微克隆技术,正向分子细胞遗传学领域迈进。生化遗传学已大步跨入分子遗传学行列。在分子代谢病的突变性质、产前基因诊断、癌基因和肿瘤抑制基因的研究、分子生物学技术的广泛应用,以至基因治疗等方面都取得可喜的成果。90年代参与了基因组计划和新的致病基因的克隆。深信我国的医学遗传学必将迅速赶上世界水平。
三、医学遗传学在现代医学中的地位
医学遗传学已经成为现代医学中一个十分活跃的领域,并迅速向医学各学科渗透。分析其原因是:
1.遗传病对人类健康的威胁日益严重。传染病得到或基本得到控制后,遗传病的相对发病率正在增长。据估计,1岁以内的死因,先天畸形占首位;活婴中有遗传病者约占24‰。约10%的孕妇流产是因为染色体异常。3%的儿童有智力发育不全,其中4/5为遗传病引起。其次,人类遗传病的病种在不断增长。
据McKusick统计,人类单基因病及异常性状,至1993年11月1日已达6457种。染色体畸变综合征在100种左右,加上异常核型近1000种。多基因病估计不少于100种。由于后者多为常见病,故人类约有1/5-1/4的人患有某种遗传病或与遗传有关的疾病。这不能不引起人们极大的关注。当然报告病种的增加,一方面是由于对遗传病认识水平的提高,对过去已存在的遗传病加以确认;但另一方面是基于研究方法的进步,从原有遗传病中分出了若干亚型。但无论如何,遗传病病种的增加仍是不容忽视的事实。
2.有些严重危害人类健康的常见病已证明与遗传因素有关。诸如肿瘤、糖尿病、动脉粥样硬化、冠心病、高血压病、精神分裂症等。过去有些不明原因的疾病,现已确证为遗传病。可以预料,随着这类疾病病因发病机制的进一步阐明,人们将从环境和遗传两个方面提出防治对策,这是一个正在开拓的广阔领域。
3.控制人口数量,提高人口质量是我国实行计划生育的基本内容。因此,应用遗传学知识和技术,提高后代健康素质是医学遗传学的一项长远目标。
四、医学遗传学的研究技术和方法
由于医学遗传学是一门边缘学科,因此它广泛地采用了形态学、生物化学、免疫学、生物统计学等研究技术。这些技术当应用于遗传学实践时得到了发展。如医学遗传学中的染色体观察技术、基因分析技术等。
医学遗传学的研究方法需针对不同的研究目的而设计。这里主要介绍一些为确定某种疾病是否有遗传因素参与而常用的方法。
1.群体筛查法采用一种或几种高效、简便并有一定准确性的方法,对某一人群进行某种遗传病或性状的普查。这种普查需在一般人群和特定人群(例如患者亲属)中进行。通过患者亲属发病率与一般人群发病率比较,从而确定该病与遗传是否有关。如果此病与遗传有关,则患者亲属发病率应高于一般人群。而且发病率还应表现为一级亲属(父母、同胞、子女)>二级亲属(祖父母、孙子女、叔舅姨姑、侄甥)>三级亲属(堂表兄妹、曾祖父母等)>一般人群。由于同一家族成员往往有相同或相似的生活环境,故在确定某病亲属患病率是否较高时,应排除环境因素影响的可能性。通常采用的方法是:①将血缘亲属与非血缘亲属加以比较。此时应该见到血缘亲属患病率高于非血缘亲属。②养子女调查,即调查患者寄养子女与养母亲生子女间患病率的差异。例如精神分裂症女性患者生育子女后,常寄养他人家中。Heston和Denny调查了寄养子女和非寄养子女精神病及有关疾病发病情况,表明两者之间有显著差异(表1-1)。
表1-1 精神分裂症母亲的寄养子女与非寄养子女
| 非寄养子女(对照组) | 寄养子女(观察组) | |
| 子女人数 | 50(平均36.3岁) | 47(平均35.8岁) |
| 精神分裂症 | 5 | |
| 精神缺陷 | 4 | |
| 病态人格 | 2 | 9 |
| 神经官能症 | 7 | 13 |
| 住精神病院或入狱1年以上 | 2 | 11 |
2.系谱分析法通常用以辨别单基因病抑或多基因病、确定遗传方式、开展遗传咨询及产前诊断、探讨遗传异质性等(参阅第二章和第九章)。
3.双生子法双生子分两种:一种称为单卵双生(同卵双生,monozygotic twin,MZ),是受精卵在第一次卵裂后,每个子细胞各发育成一个胚胎,故它们的性别相同,遗传特性及表型特征也基本相同;另一种称为双卵双生(异卵双生,dizygotic twin,DZ),来源于两个卵子分别与精子受精而发育成的两个胚胎,故其性别不一定相同,遗传特征及表型仅有某些相似。两种双生子可从外貌特征、皮纹、血型、同工酶谱、血清型、HLA型或DNA多态性加以鉴定。单卵双生子在不同环境中生长发育可以研究不同环境对表型的影响;双卵双生子在同一环境中发育生长可以研究不同基因型的表型效应。通过比较单卵双生和双卵双生某一性状(或疾病)的发生一致性(concordance),可以估计该性状(或疾病)发生中遗传因素所起作用的大小。一般可用发病一致率(同病率)来表示。
发病一致率(%)=同病双生子对数/总双生子(单卵或双卵)对数×100
例如结核病MZ同病率为74%,DZ同病率为28%,可以认为结核病的发生有一定遗传背景。表1-2列举了几种双生子法研究几种与遗传因素有关疾病的结果(其中麻疹与遗传因素关系较小)。
表1-2 几种疾病单卵双生子与双卵双生子发病一致率的比较
| 疾 病 | 发病一致率(%)单卵双生 | 双卵双生 |
| 先天愚型 | 89 | 7 |
| 精神分裂症 | 80 | 13 |
| 结核病 | 74 | 28 |
| 糖尿病 | 84 | 37 |
| 原发性癫痫 | 72 | 15 |
| 十二指肠溃疡 | 50 | 14 |
| 麻疹 | 95 | 87 |
4.种族差异比较种族是在繁殖上隔离的群体,也是在地理和文化上相对隔离的人群。各个种族的基因库(群体中包含的总的遗传信息)彼此不同。世界上主要的人种有6种,即高加索人(白种人)、黑种人、亚洲蒙古种人、美洲印第安人、澳大利亚种人及巴斯克人(西班牙及法国南郊)。每一种还可分为若干亚种。种族的差异具有遗传学基础。不同种族的肤色、发型、发色、虹蟆颜色、颧骨外形、身材等外部形态性状都显示出遗传学差异。它们之间在血型、组织相容性抗原(HLA)类型、血清型、同工酶谱等的基因型频率也不相同。因此,如果某种疾病在不同种族中的发病率、临床表现、发病年龄和性别、合并症有显著差异,则应考虑该病与遗传密切有关。例如中国人的鼻咽癌发病率在世界上居首位。在中国出生侨居美国的华侨鼻咽癌发病率比当地美国人高34倍。当然,不同种族生活的地理环境、气候条件、饮食习惯、社会经济状况等方面也各不相同,故在调查不同种族发病率及发病情况时,应严格排除这类环境因素的影响。为此,这种调查常安排在不同种族居民混杂居住的地区进行,最好选择生活习惯和经济条件比较接近的对象。
5.疾病组分分析疾病组分分析(component analysis)是指对待比较复杂的疾病,特别是其发病机制未完全弄清的疾病,如果需要研究其遗传因素,可以将疾病“拆开”来对其某一发病环节(组分)进行单独的遗传学研究。这种研究方法又称为亚临床标记(subclinical marker)研究。如果证明所研究的疾病组分受遗传控制,则可认为这种疾病也有遗传因素控制。
6.伴随性状研究在疾病的研究中,如果某一疾病经常伴随另一已确定由遗传决定的性状或疾病出现,则说明该病与遗传有关。性状的伴随出现可以是由于基因连锁(link-age),即两个基因座位同在一条染色体上;也可以是由于关联(association),即两种遗传上无关的性状非随机的同时出现。属于连锁的伴随性状如椭圆形红细胞增多症常见于Rh血型阳性者。现知这两种性状的基因紧密连锁,前者定位于1号染色体短臂3区2带(1p32);后者定位于1p35。属于关联的伴随性状如O型血者十二指肠溃疡发病率较其他血型高30%-40%。较多用的遗传标记为HLA(humanleucocyte antigen)系统。HLA包括7个连锁座位(A,B,C,D,DR,DP,DQ)148个复等位基因。例如强直性脊柱炎与HLA系统的B27等位基因有关联。强直性脊柱炎患者中HLA-B27频率非常高,相对风险比为87.4,即有B27者,患本病风险是无B27者的87.4倍。最近已广泛使用DNA多态遗传标记(详见第四章、第十三章)。有时,已确定的遗传病也可作为遗传标记,如溃疡性结肠炎常与强直性脊柱炎伴发,由于后者已证明为遗传病,故前者可认为与遗传因素有关。
7.动物模型由于直接研究人类遗传病受到某些限制,故动物中存在的自发遗传病可以作为研究人类遗传病的辅助手段。但应注意所得结论仅可作参考,但不能搬用于人类。近年来研究成功的转基因动物,特别是转基因小鼠,已有人工定向复制可传代的动物模型,大大的丰富了这一手段。
五、遗传性疾病概述
(一)遗传因素在疾病发生中的作用
人们对疾病有着不同的认识。因此疾病也有各种各样的定义。遗传学家往往认为形态或代谢异常的性状就是疾病;临床学家则认为疾病是有特定症状和体征的病态过程;生物学家将疾病看成是内环境稳态的失衡。从环境与机体统一的观点看,疾病是环境因素(外因)和机体(内因)相互作用而形成的一种特殊的生命过程,伴有组织器官形态、代谢和(或)功能的改变。遗传因素是构成内因的主要因素。因此,可以认为,任何疾病的发生都是环境因素与遗传因素相互作用的结果。但在某一具体疾病发生中,环境因素与遗传因素的相对重要性则要具体分析。大致有下面三种情况:第一类是环境因素起主要作用的疾病。第二类是遗传因素起主导作用的疾病。第三类是环境因素与遗传因素都很重要,遗传因素提供了产生疾病的必要的遗传背景,环境因素促使疾病表现出相应的症状和体征(图1-1)。但三者之间并无严格的界限,例如维生素C缺乏症(坏血病)是环境因素起主导作用的疾病。这是因为人类普遍缺乏体内合成维生素c 必需的古洛酸糖内酯氧化酶,所以必需外源性维生素C,因此维生素C缺乏症也可看成是此酶遗传性缺乏的结果。任何表现型都是基因型与环境相互作用的结果,遗传因素起主导作用的疾病,也都有环境因素参与。
(二)遗传性疾病的概念
遗传性疾病(hereditary disease ,inherited disease,genetic disease)简称遗传病,是指生殖细胞或受精卵的遗传物质(染色体和基因)发生突变(或畸变)所引起的疾病,通常具有垂直传递(vertical transmission)的特征。
上述定义强调了遗传学病的三个方面:①垂直传递。遗传病不同于传染病的水平传递(horizontaltransmission),而是具有上代往下代传递的特点。但不是每个遗传病的家系中都可观察到这一现象。因为有的患者是首次突变产生的病例。是家系中的首例;有些遗传病特别是染色体异常的患者,由于活不到生育年龄或不育,以致观察不到垂直传递的现象。②遗传物质(主要是指基因,由于染色体是基因的载体,为叙述方便,也将染色体归入这一概念)的突变(或染色体畸变)。这是遗传的物质基础。③不是任何细胞的遗传物质改变都可以传给下代,所以必须强调生殖细胞或受精卵的遗传物质发生改变。例如人在遭受电离辐射后可以产生放射病,此时,皮肤细胞、骨髓细胞等体细胞的遗传物质可以发生改变,但放射病不能传给下一代。所以这里必须强调生殖细胞或受精卵中的遗传物质发生改变,如果体细胞遗传物质突变传给了子细胞,这种情况可以认为是一种体细胞遗传病,有人将肿瘤看成是一种体细胞遗传病。
遗传病不应与先天性疾病(congenital disease)等同看待。先天性疾病是指个体出生后即表现出来的疾病。如果主要表现为形态结构异常,则称为先天畸形(congenital anomaly)。应该指出,许多遗传病在出生后即可见到,因此大多数先天性疾病实际上是遗传病,但也有某些先天性疾病是在子宫中获得的,如风疹病毒感染引起的某些先天性心脏病,药物引起的畸形等。反之,有些出生时未表现出来的疾病,也可以是遗传病。如原发性血色病(primary hematochromatosis)是一种铁代谢障碍疾病,但铁要积存到15g以上才发病,故80%病例发病年龄在40岁以上。
据估计,先天性疾病中,已肯定主要为遗传因素引起的仅占10%左右,主要在子宫中或产程中后天获得的也仅约占10%,尚不能分清(包括遗传与环境因素共同作用)的约占80%,现已有专门研究先天畸形的学科,称为畸形学(dysmorphology)。在实际工作中,要清楚区分哪些是遗传的,哪些是在发育中获得的,往往很困难。下列几点有助于判断两种情况:
1.产妇在妊娠过程中,特别是妊娠的15-60天内(高敏感期)有否接触过致畸因素,又称致畸剂(teratogen)。1962年,西德发现在出生婴儿中有大批(10,000例以上)无肢或短肢畸形。据分析可能是由于孕妇因妊娠反应服用了当时新合成的一种抗恶心和安眠药——酞胺哌啶酮(thalidomide,又名反应停)。后来,当局下令停止生产和出售该药,该类畸形很快匿迹。
2.通过了解生产史,估计环境影响胎儿发育的可能性。例如先天性髋脱位的婴儿多有臀位生产史,表明髋脱位是胎位不正所致。
3.先天畸形的发病率如果随季节、时间、地理或社会经济等环境条件而变化,则这种畸形多为环境因素引起。例如,最初怀疑酞胺哌啶酮致畸就是从发病率的突然变化而察觉。
遗传病也与家族性疾病(familial disease)加以区别。家族性疾病是指某种表现出家族聚集现象的疾病,即在一个家庭中不止一个成员罹患。当然,许多遗传病(特别是显性遗传病)常见家族聚集现象,但也有不少遗传病(特别是隐性遗传病和染色体病)并不一定有家族史。故“家族性”一词一般用于表达未弄清病因而又怀疑可能为遗传病时,但在弄清病因后,应该代之以“遗传性”。然而,由于习惯,至今仍沿用家庭性高胆固醇血症、家族性甲状腺肿等名称。
六、遗传病的分类及发病率
遗传病一般分为基因病(genicdisease)与染色体病(chromosomal disease)。基因病又分为单基因病(monogenic disease)和多基因病(polygenic disease)。单基因病可按遗传方式而细分;染色体病可按常染色体和性染色体异常分两大类(表1-3)。
表1-3 遗传病的分类及发病率
| 分 类 | 发病率(%) |
| 基因病 | |
| 单基因病 | 2.5 |
| 常染色体显性遗传病 | 0.9 |
| 常染色体隐性遗传病 | 1.3 |
| 性连锁遗传病 | 0.3 |
| 多基因病 | 18.0 |
| 染色体病 | 0.54 |
| 常染色体异常 | 0.36 |
| 性染色体异常 | 0.182 |
单基因病未计红绿色盲及地区性发病如G6PD缺乏症和地中海贫血等
遗传病的发病率目前还缺乏确切的资料,这是由于诊断水平、研究方法、群体选择以及分类标准不同,各地区不同学者报告有较大差别。在我国还缺乏全面的调查和确诊的手段。表1-3中列出的数字仅为一粗略估计,其中染色体病的发病率比较准确。
第二章 染色体病
第一节 人体染色体
一、人体染色体数目、结构和形态
人类体细胞具有46条染色体,其中44条(22对)为常染色体,另两条与性别分化有关,为性染色体。性染色体在女性为XX,在男性为XY。生殖细胞中卵细胞和精子各有23条染色体,分别为22+X和22+Y。
染色体在细胞周期中经历着凝缩(condensation)和舒展的周期性变化。在细胞分裂中期,染色体达到凝缩的高峰,轮廓结构清楚,因而最有利于观察(图2-1)。
每一中期染色体都由两条染色单体构成,它们各含一条DNA双螺旋链。两条单体仅在着丝粒外互相连接,该处为染色体的缩窄处,故又称为主缢痕。着丝粒是纺锤丝附着之点,在细胞分裂中染色体的运动密切相关,失去着丝粒的染色体片段通常不能在分裂后期向两极移动而丢失,着比粒又将染色体横向地分为两个臂。图2-1为中期染色体的模式图。
根据着丝粒的位置,人类染色体可以分为三种:①近中着丝粒染色体,着丝粒位于或靠近染色体中央,将染色体分为长短相近的两个臂;②亚中着丝粒染色体,着丝粒偏于一端,将染色体分为长短明显不同的两个臂;③端着丝粒染色体,着丝粒靠近一端,人类没有真正的端着丝粒染色体(图2-1)。

图2-1 中期染体模式图
右图;人类三种染色体:近中(a)、
亚中(b)、及端(c)着丝粒染色体
二、核型和分组
任何一条染色体重要的形态特征是着丝料的位置和相对长度。着丝粒将染色体分为短臂(以p表示)和长臂(以q表示)。着丝粒的位置可在显微镜下直接观察,精确测量。
将一个细胞内的染色体按照一定的顺序排列起来所构成的图像称为该细胞的核型(karyotype)。通常是将显微镜摄影得到的染色体照片剪贴而成(图2-2)。一个细胞的核型一般可代表该个体的核型。核型如用模式图表示则称为组型(idiogram)。
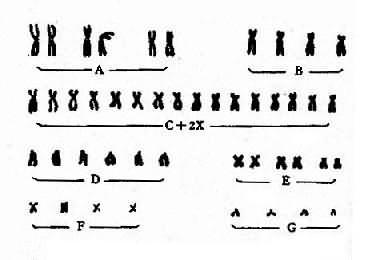
图2-2 一个女性的未显带核型
早期,根据染色体的长度和着丝粒位置将人类染色体顺次由1编到22号,并分为A、B、C、D、E、F、G7个组。将X和Y染色体分别归入C组和G组(图2-2)。但据此要准确鉴别多数组内染色体的序号是困难的。
1.显带染色体 70年代初,瑞典细胞化学家Caspersson首先应用荧光染料喹吖因氮芥(quinacrine mustard)处理染色体标本,发现在荧光显微镜下每条染色体出现了宽窄和亮度不同的纹,即荧光带,而各条染色体有其独特的带型,由此可以清楚地鉴别人类的每一条染色体。
用此法显带称Q显带。后来发现将染色体标本用热、碱、胰酶、尿素、去垢剂或某些盐溶预先处理,再用Giemsa染料染色,也可以显示类似带纹,称为G显带(图2-3)。用其它方法还可以得到与G带明暗相反的R带(reverse bands)和专门显示着丝粒异染色质的C带,以及专一显示染色体的端粒(T显带)或核仁组织区 (N带)和各种带型。

图2-3 一个男性G显带中中期分裂象
显带技术不仅解决了染色体的识别问题,由于染色体上能区别许多区和带,还为深入研究染色体的异常和人类基因定位创造了条件。
显带染色体模式图和命名:为了例于交流,1971年召开的巴黎会议曾制订了一幅显带染色体模式图并对命名作了详细的规定(图2-4)。由图可见,每条染色体仍以数字编号并分为短臂(p)和长臂(q),每条臂又分为若干区和带,次递以数字表示,如3p14代表3号染色体短臂1区4带。在此模式图的基础
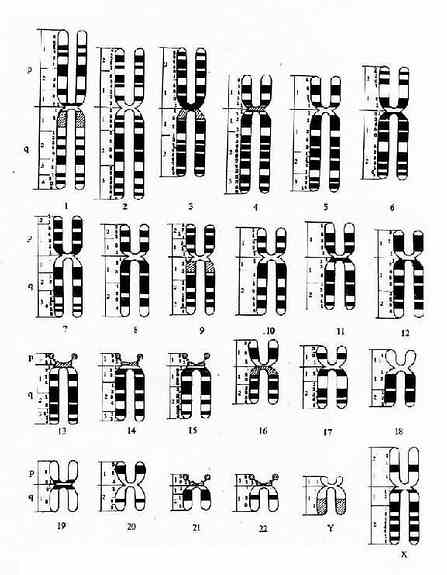
图2-4 显带染色体模式图(巴黎会议,1971)
上以后又制订了人类细胞遗传学命名的国际体制(ISCN,1978),并几经修改。
2.高分辩显带染色体 巴黎会议模式图中,一套单位染色体共有332条带。70年代后期,由于细胞同步化方法的应用和显带技术的改进,人们已能得到更长和带纹更加丰富的染色体,这种染色体称为高分辩显带染色体(high resolution banding)。它能提供染色体及其畸变的更多细节,有助于发现更多细微的染色体异常,使染色体结构畸变的断点定位更加准确,因而在临床细胞遗传学检查或肿瘤染色体研究以及人类基因定位中被采用。
第二节 人体染色体畸变
一、染色体的数目畸变
正常人的体细胞具有46条染色体(2n),配子细胞(精子和卵)具有23条染色(n),前者称为二倍体,后者称为单位体。染色偏离正常数目称为染色体数目异常或数目畸变。
1.多倍体和多倍性体细胞染色体倍数超过2倍,即是3n=69,4n=92等时,这些细胞称为多倍体细胞,而这种状态称为多倍性(polyploidy)。在人类,全身三倍性是致死的,因而极为罕见,但三倍性在流产胎儿中较常见,是流产的重要原因之一。全身三倍性可能是由于参加受精卵细胞为二倍体而非单倍体,或由于双精子受精所致。
全身四倍性更多罕。但四倍体和其它高倍细胞在一些组织发肝、子宫内膜、骨髓细胞、瘤组织和培养细胞中并不罕见。其产生的原因是,如果细胞在分裂之前再复制一次,或由于纺锤体的缺陷或缺如,细胞未能分裂,都会使染色体数目倍增。
2.异倍性或非整倍性(aneupoloidy)细胞的染色体数不是23的整倍时,称为异倍体细胞,如细胞具有44,45,48,67,90条染色体时都是异倍体细胞,44和45略少于46,故可称为亚二倍体;47,48略多于46,称为超二倍体。同理,67可称为亚三倍体等。异倍体细胞在肿瘤组织十分常见。发生的原因是染色的丢失,某些染色体的核内复制(endoredplication)或染色体的不分离。
3.三体性和单体性体细胞在减数分裂时如发生某号染色不分离,则导致该染色体增多一条(三体性,trisomy)或减少一条(单体性,monosomy)。除21、13、18、和22三体性外,其它三体性多导致流产(嵌合状态者除外,如嵌合性的8、9、10号三体性等)。性染色体三体性常见一些。常染色体的单体性严重破坏基因平衡,因而是致死的。但X染色体单体的女性还可见于儿童或成人。(表2-1)
表2-1 1863例染色体异常的自发流产儿中各种异常的频率
| 染色体异常 | 频率(%) |
| 三全性 | 52 |
| 45,X | 18 |
| 三倍性 | 17 |
| 四倍性 | 6 |
| 其它(主要是易位) | 7 |
染色体数目异常的机理:在细胞分裂时,如果某一染色体的两条单体在分开后的期不能正常地分开而同时进入某一子细胞,则必然导致该子细胞增多一条染色体而另一子细胞缺少一条染色体,这称为染色体不分离(nondisjunction)。
如不分离发生在减数分裂,所形成的异常配子与正常配子结合后,就会出现合子细胞中某一染色体的三体性或单体性。不分离可以发生在第一次减数分裂,也可以发生在第二次减数分开。不分离产生的异常配子在受精后导致合子染色体异常,因此由合子分裂得来的全身细胞都具有该种异常(图2-5)。
另一情况是,合子细胞最初是正常的,但在以后的某次有丝分裂时发生不分离,这也能导致染色数目异常。这种异常细胞如能存活和继续分裂,将构成异常的细胞系,并与正常细胞系并存。具有染色体组成不同的两种或两种以上细胞系的个体称为嵌合体(mosaic)(图2-5)。
还有一种造成个别染色体数目异常的原因是染色体丢失(chromosome loss)。这是由于有丝分裂后期染色单体的迟留(anaphaselag)所致。导致本应向子细胞移动的某一染色体(此时为单体状态)未能与其它染色体一起移动而进入了细胞,并随后丢失,这就导致某一子细胞及其后代中该染色体减少一条。(图2-6)。
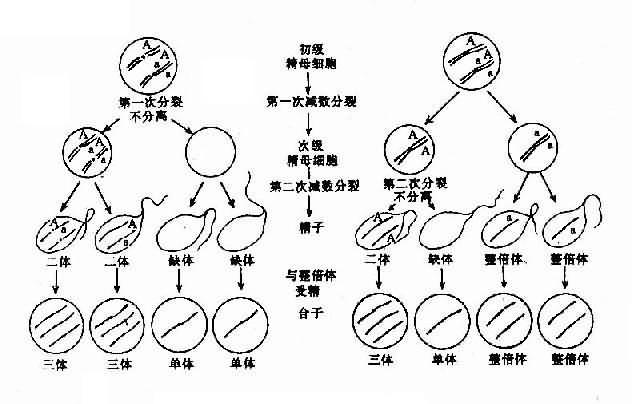
图2-5 减数分裂时染色体不分离
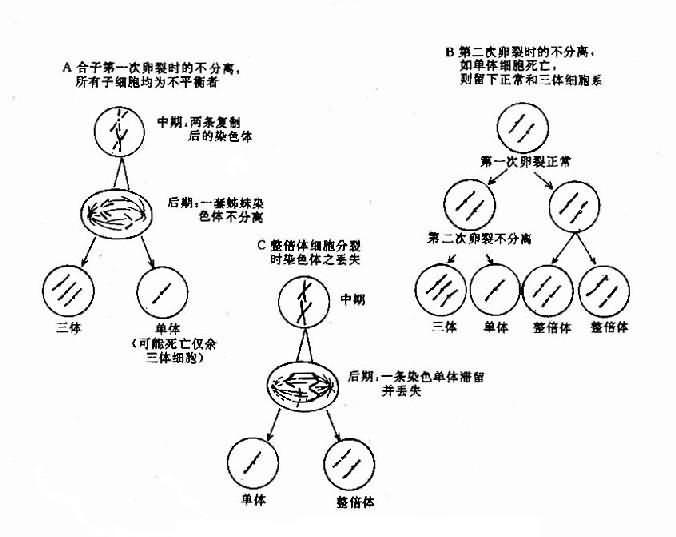
图2-6 嵌合体形成的机理示意图有丝分裂不分离(A,B)及染色体的丢失(C)
二、染色体的结构异常
许多物理、化学和生物因子可以引起染色体断裂(breakage),这些因子称为致断因子(clastogenic factor)或染色体断裂剂。此外,染色体也能自发断裂。断裂端被认为具有“粘性”,即易与其它断端接合或重连(reunion)。因此,一次断裂产生的两个粘性末端通常重连而修复如初。但有时出现非正常的重连,结果导致多种染色体结构异常。
根据断裂发生时染色体是否已复制,结构异常可分为两大类型:即染色体型和单体型。如断袭发生于G1期,即染色体尚未复制而只有一条单体,断裂通过S期时的复制而影响到两条单体,将导致染色体型的异常。如断裂发生在G2期,此时染色体已完成复制,由两条单体组成,断裂通常只涉及其中一条单体,导致单体型结构异常。以下着重叙述染色体型结构常。
常见的染色体型结构异常有以下几种:
1.缺失染色体部分丢失称为缺失(deletion,用del表示)(图2-7)。当一条染色体发生两次断裂,其间的片段丢失,称为中间缺失(intersititial deletion)。虽然缺失是中间缺失,但在显微镜下像是末端缺失。
2.环状染色体当一条染色体的两臂各有一次断裂,有着丝粒节段的两个断端如彼此重新连接,可形成环状染色体(ring chromosome,用r表示)(图2-7)。这在辐射损伤时尤为常见。
3.等臂染色体一次染色体断裂如果发生在着丝粒区,使着丝粒横断,则两个臂的姐妹染色单体可分别互相连接,结果形成两条与短臂和长臂相应的等臂染色体(isochromosome,用i表示)(图2-8)。当然,等臂染色体还可能有其它的形成机理,如通过两条同源染色体着丝粒融合,然后短臂和长臂分开,两条短臂和两条长臂借着丝料分别各自连接成一条等臂
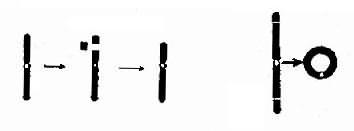
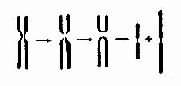
图2-7 染色体缺失及环状染色体的形成
图2-8等臂染色的形成
左图:中间缺失 右图:环状染色体形成染色体。
4.倒位如果两次断裂形成的片段倒转180度重新接合,那么,虽然没有染色体物质的丢失,但基因顺序颠倒,称为倒位(inversion,用inv表示)。如果倒位发生在同一臂内,称为臂内倒位(paracentric inversion);如果两次断裂分别发生在长臂和短臂,则称为臂间倒位(paracentric inversion)。在应用显带技术以前,臂内倒位是无法检出的,因为染色体的长度和臂率(p/q长度比)都没有改变。至于臂间倒位,如果两断点距着丝粒不等,则能被发现(图2-9)。倒位因无染色体物质的增减,一般没有明显的表型效应。
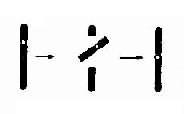
图2-9 染色体的臂间例位
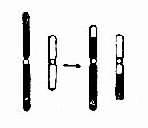
图2-10 染色体相互易位示意图
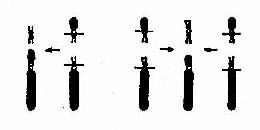
图2-11 罗氏易位的机理及结果
5.易位染色体片段位置的改变称为易位(translocation,用t表示)。它伴有基因位置的改变。易位发生在一条染色体内时称为移位(shift)或染色体内易位(intrachromosomal translocation);易位发生在两条同源或非同源染色体之间时称为染色体间易位(intrachromosomal translocation)。染色体间的易位可分为转位(transposition)和相互易位(reciprcal translocation,用rcp表示)。前者指一条染色体的某一片段转移到了另一条染色体上,而后者则指两条染色体间相互交换了片段。
(1)相互易位:两条染色体发生断裂后相互交换无着丝粒断片形成两条新的衍生染色体为相互易位(图2-10)。相互易位是比较常见的结构畸变,在各号染色体间都可发生,新生儿的发生频率约1-2/1000。相互易位仅有位置的改变,没有可见的染色体片段的增减时称为平衡易位(balanced translocation)。它通常没有明显的遗传效应。然而平衡易位的携带者与正常人婚后生育的子女中,却有可能得到一条衍生异常染色体,导致某一易位节段的增多(部分三体性)或减少(部分单体性),并产生相应的效应。
(2)罗氏易位:罗氏易位(Robertsonian translocation)为相互易位的一种特殊形式。两条近端着丝粒染色体(D/D,D/G,G/G)在着丝粒处或其附近断裂后形成两条衍生染色体。一条由两者的长臂构成,几乎具有全部遗传物质;而另一条由两者的短臂构成(图2-11),由两个短臂构成的小染色体。由于缺乏着丝粒或因几乎全由异染色质组成,故常丢失。它的存在与否不引起表型异常。
罗氏易位通常又称为着丝粒融合(centricfusion)。在减数分裂时,由于由两条短臂构成的小染色体丢失,故在联会时只有三条染色体参与,形成三价体(trivalent)(图2-12)。三价体的分离方式有三种,即交替式(同源着比粒各走向一极,结果产生一种正常的和一种平衡易位的配子)、邻式-1和邻式-2。同源着丝粒均走向一级,亦即易位染色体与某一条正常染色体同走向一极,结果均形成二体(重复)或缺体的配子。这种配子在受精后形成三体性或单体性的合子。由于缺体的配子通常是致死的,因而实际上可能参与受精的配子只有三种:正常的、带有平衡易位的和导致三体性的配子。罗氏易位的携带者尽管只有45条染色体,但除偶有男性不育外,没有表型异常。这是因为易位染色体几乎包括了两条长臂的全部,没有基因的大量丢失,而丢失了两条短臂几乎全是结构异染色质。
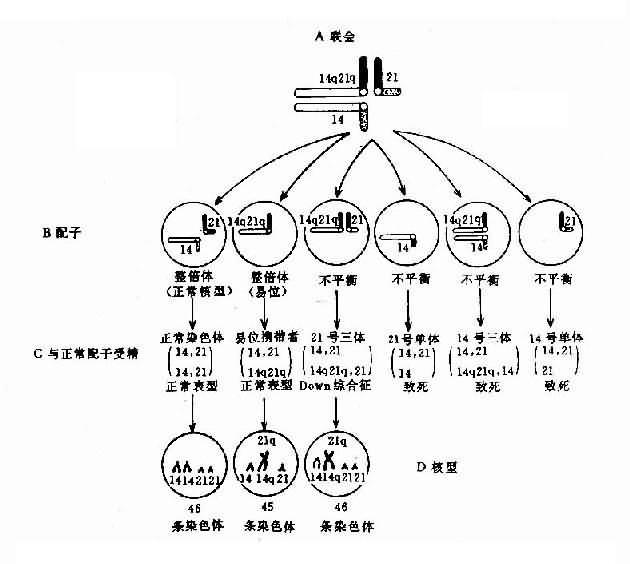
图1-12罗氏易位(14q21q)杂合体减数分裂时染色体的联会
(A)、分离(B)、正常配受子精结果(C)及某些具有代表性的核型(D)
6.双着丝粒染色体两条染色体断裂后,具有着丝粒两个片段相连接,即形成一个双着丝粒染色体(图2-3)。两个无着丝粒片段也可以连接成一个无着丝粒片段,但后者通常在细胞分裂时丢失。双着丝粒染色体常见于电离辐射后,因此在辐射遗传学中常用以估算受照射的剂量。
7.插入一条染色体的某一节段插入另一染色体中称为插入(insertion,用ins表示)。显然,只有发生了三次断裂时插入才有可能(图2-14)。插入可以是正位的,也可以是倒转180度后反向的。插入如发生在同源染色体间,则导致一条染色体中发生重复,而另一条同源染色体中发生同一节段的缺失。
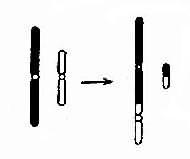
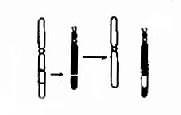
图2-13 双着丝粒染色体的形成 图2-14染色体插入示意图
8.重复染色体上个别区段多出一份,称为重复(duplication,用dup表示)。除相互易位外,插入也是导致重复的主要原因。例如,一条染色体两次断裂后,其中一条单体的断片可以手稿另一单体的任一断口。在细胞分裂后,一条染色体缺失了两个断口之间的节段,而另一染色体却有该节段的重复(图2-15)。类似的插入也可发生在减数分裂过程中两条同源染色体间,造成全身性的重复和缺失。
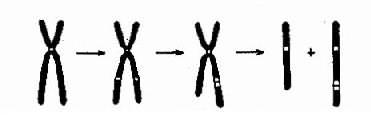
图2-15 染色体重复示意图
三、核型的描述
核型的描述,首先是书写染色体总数,加一个逗号,接着写出性染色体的组成,然后写出染色体的异常。一些常用符号的含义见表2-2。
表2-2 常用符号及其意义
| ace | 无着丝粒片段 | r | 环状染色体 |
| cen | 着丝粒 | rcp | 相互易位 |
| del | 缺失 | rea | 重排 |
| der | 衍生染色体 | rob | 罗氏易位 |
| dic | 双着丝粒染色体 | : | 断裂 |
| dup | 重复 | ∷ | 断裂后重接 |
| h | 次缢痕 | () | 括号内为结构异常的染色体 |
| i | 等臂染色体 | ; | 重排中用于分开染色体 |
| ins | 插入 | / | 嵌合体中用于分开不同的细胞系 |
| inv | 倒位 | t | 易位 |
| p | 短臂 | ter | 末端 |
| q | 长臂 | 从....到 |
“+”和“-”号当其放在相应的符号之前,表示增加或丢失了整条染色体;当其放在相应符号之后,则表示染色体长度的增加或减少。例如:47,XX,+21为一个女性先于愚型的核型,有一条额外的21号染色体;46,XY,5p-表示一个5号染色体短臂长度减少的男性核型。
结构异常染色体的核型描述可用简明或详尽描述系统来表示,前者指出了异常核型,并可推论出异常染色体的带的构成;后者除指出重排类型外,学依据其带的构成说明了每一条异常染色体,现举数例如下:
1.末端缺失
46,XX,del (1) (q21)
46, XX,del (1) (pter→ q21: )
表示1号染色体长臂2区1带处断裂造成了该处以远的末端缺失,异常的染色体由完整的短臂和着丝粒与1q21带之间的部分长臂构成。
2.中间缺失
46,XX,del (1) (q21q23)
46, XX,del (1) (pter →q21∷q31→qter )
表示1号染色体长臂2区1带与1区1带处断裂,其间片段丢失,下行括号内说明了异常染色体的构成。
3.臂间倒位
46,XY,inv (2) (p21q31)
46, XY, inv(2) (pter→ p21∷q31→p21∷q31→qter )
断裂和重接在2号染色体短臂的2区1带和长臂的3区1带之间,其间的节段倒置。
4.环形染色体
46,XY, r (2) (p21q31)
46, XY, r (2) (p21→ q31 )
5、相互易位
46,XY,t (2;5) (q21;q31)
46,XY, t (2;5) (2pter→ 2qter ∷5q31→5qter;5pter→ 5q31∷2q21→2qter)
断裂和重接分别发生在2号染色体和5号染色体长臂的2q21和5q31带,这些带以远的节段在两条染色体之间进行了交换,后面括号内描述小号数的衍生染色体(本例为2号)
6.等染色体
46,X, i (Xq)
46, X, i (X) (qter→ cen→ qter)
女性核型,有一条正常的X染色体和一条X染色体长臂形成的等臂染色体。
四、姐妹染色单体交换
一条染色体的两条单体在同一位置发生同源片段的变换,称为姐妹染色单体交换(sister chromatid exchange,SCE)。由于交换是对等的,所以染色体的形成没有改变,但用特殊的培养液和处理方法可以显示出来(图2-16)。
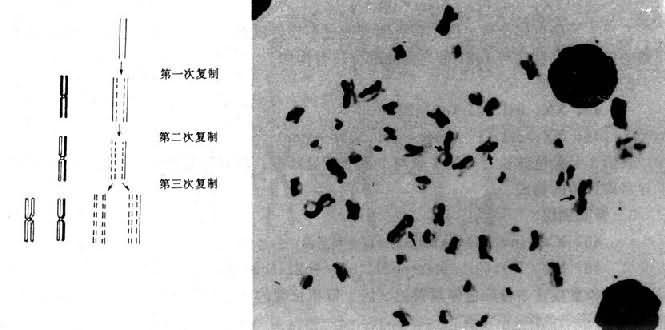
图2-16 姐妹染色体单位交换
左图:姐妹染色单体区染色原理(虚线为Brdu取代后的DNA链)
右图:姐妹染色单体交换(箭头示交换部位)
SCE的遗传学意义还不完全清楚,是否存在自发的SCE也还有争议,交换的机理尚未完全阐明,但它显然与DNA损伤和修复过程有关。作为一种简便和敏感的遗传学指标,它在诱变和肿瘤研究等领域中的应用十分广泛。例如,目前已知许多环境诱变剂、职业有害因素、抗肿瘤药物、病毒等都可以引起SCE率增加,Bloom综合征患者和某些肿瘤患者的SCE率也明显上升。
第三节 染色体畸变综合征
染色体病(chromosomaldisease)或染色体畸变综合征(chromosome aberration syndrome )是一大类严重的遗传病,通常伴有发育畸形和智力低下,同时也是导致流产与不育的重要原因。一般估计染色体畸变见于0.5%-0.7%的活产婴儿,7.5%的胎儿,自发流产儿约1/2有染色体异常。现今已知的染色体病超过100种,已报告的染色体数目和结构异常在500种以上。随着高分辩显带及其它细胞遗传学新技术的应用,今后还会发现更多的染色体病和异常。
一、染色体畸变综合征的概念。
染色体畸变综合征是指由于染色体异常而引起的疾病。由于它有多种临床表现,故称为综合征。通常如果没有染色体物质明显增多或减少。如一些染色体重排(平衡易位、倒位)就不一定引起表型异常。染色体的多态或异态性(polymorphism或heteromorphism)通常不伴有异常表型,故不称为染色体畸变综合征。
二、染色体异常发生的频率
综合许多国家的资料,大约有15%的妊娠发生流产,而其中一半为染色体异常所致,即约为5%-8%的胚胎有染色体异常。不过在出生前,90%以上已有自然流产或死产。流产愈早,有染色体异常的频率愈高。新生儿染色体异常调查结果见表2-3。
不同地区染色体异常发生的频度相关不大,波动于0.47-0.84%之间,用表2-3中的发病率对我国新生儿中染色体异常发病率作了外推估算(表2-4)。
普通成人染色体调查的资料很少。1986-1987年,我国四川省曾进行过大规模的遗传病流行病学抽样调查,其染色体病患者的患病率如表2-5。
表2-3 56 952名新生儿细胞遗传学检查结果
| 染色体异常类型 | 异常人数 | % | 近似发病率 |
| 性染色体-男性 | 98 | 0.260 | 1/400 |
| 47,XXY | 35 | 0.093 | 1/1 000 |
| 47,XYY | 35 | 0.093 | 1/1 000 |
| 其它 | 28 | 0.074 | 1/1 300 |
| 性染色体-女性 | 29 | 0.151 | 1/700 |
| 45,X | 2 | 0.010 | 1/10 000 |
| 47,XXX | 20 | 0.014 | 1/1 000 |
| 其它 | 7 | 0.037 | 1/3 000 |
| 常染色体三体性 | 82 | 0.144 | 1/700 |
| +D | 3 | 0.005 | /20 000 |
| + E | 7 | 0.120 | 1/8 000 |
| + G | 71 | 0.125 | 1/800 |
| 其它 | 1 | 0.002 | 1/50 000 |
| 平衡的结构重排 | 110 | 0.193 | 1/500 |
| 不平衡结构重排 | 34 | 0.597 | 1/2 000 |
| 总计 | 353 | 0.620 | 1/160 |
表2-4 我国新生儿染色体异常人数的估算
| 异常类型 | 异常频率% | 外推值(名/年) |
| 性染色体异常 | 0.223 | 46138 |
| 常染色体数目异常 | 0.144 | 29793 |
| 染色体结构重排 | ||
| 平衡的 | 0.193 | 39793 |
| 不平衡的 | 0.060 | 12414 |
| 总计 | 0.620 | 128276 |
(按1981年出生人口外推计算)
表2-5 四川省普通人群中染色体病的患病率
| 病名 | 患病率 |
| 21-三体性 | 0.14 |
| 其它常染色体病 | 0.02 |
| 先天性卵巢发育不全 | 0.07 |
| 先天性睾丸发育不全 | 0.07 |
| 其它染色体异常 | 0.015 |
| 总计 | 0.315 |
*先天性睾丸发育不全,可能因为筛查困难而数值偏低
三、常染色体异常综合证
(一)三体综合征
1.先天愚型先天愚型是最重要的染色体疾病。英国医生Langdon Down 首先描述,故称为 Down综合征(Down sydrome)。1959年,法国细胞遗传学家Lejeune证实此病的病因是多了一个小的G组染色体(后来确定为21号),故此病又称为21三体综合征。Lejeune的发现开创了医学遗传学的一个重要分支――临床细胞遗传学。
(1)发病率:新生儿中21三体综合征的发病率约为1/800或1.25%,但男性患儿多于女性。母亲年龄是影响发病率的重要因素。根据国外资料,如果一般人出生时的母亲年龄平均28.2岁,则本病患儿母亲平均年龄为34.4岁,如母龄20岁后为1:2000,35岁后为1:300,40岁后为1:100,45岁后升至1:50。
(2)临床表现:先天愚型患儿出生时体重和身长偏低,肌张力低下,突出的是颅面部畸形(图2-17),头颅小而圆,枕部扁平,脸圆而扁平,鼻扁平,脸裂细且上外倾斜,眼距过宽,内眦赘皮明显,常有斜视,虹膜时有白斑点,常见晶状体混浊,嘴小唇厚,舌大外伸(伸舌样痴呆之名由此而来),耳小,耳位低,耳廓畸形,颈背部短而宽,有多余的皮肤,由于软骨发育差,患者四肢较短,手宽而肥,通贯掌,指短,第5指常内弯,短小或缺小指中节,皮纹也有一定的特点(参阅第十章),腹肌张力低下而膨胀,故常有腹直肌分离或脐疝,约1/2以上的患者有先天性心脏病,主要是室间隔缺损、房室道连通、和运脉导管未闭,消化道的畸形如十二指肠的狭窄、巨结肠、直肠脱垂及肛门闭锁等也偶尔可见。在男性常有隐睾,睾丸有生精过程,但精子常减少,性欲下降,尚未见有生育者。女性患者通常无月经,但有少数能妊娠和生育。精神发育迟滞或智力低下(mental retardation,MR)是本病最突出最严重的表现,但其程度在各患者不完全相同,智商通常在25-50之间,高于50的很少。行为、动作倾向于定型化,抽象思维能力受损最大。
(3)实验室检查:过氧化物岐化酶(SOD-1)活性可增高50%,该酶基因定位21q22,即具有基因剂量效应。此外,患者对阿托品特别敏感,患者乙酰胆碱缺陷或许可以解释智力低下、应变力差、便秘等症状,而免疫功能失调,如淋巴细胞和丙球蛋白减少则是患儿易感染的原因。
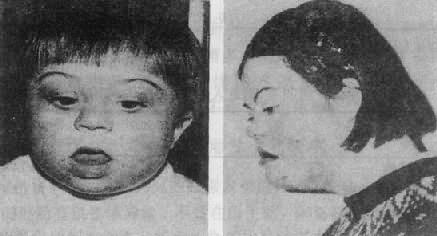
图2-17 先天愚型患者
右图:1岁女孩
左图:16岁女孩
(4)核型:核型可分为三型,各型的比例是:典型的(游离型),即47,+21占95%;嵌合型即46/47,+21占1%-2%;易位型占3%-4%。游离型全身体细胞均多一条21号染色体,临床症状典型而且显著。由于嵌合型通常具有两个细胞系,其症状表现取决于异常细胞所占的比例,故差异很大,但一般较典型者为轻。如果三体细胞很少,则表现与正常人无异。易位型的核型有多种,最常见的是Dq21q,占全部易位型的54.2%,其次是21qGq,占40.9%,其它易位型5%。一般说来,易位型的症状比典型的要轻些,在Dq21q中,最常见的是14q21q,占Dq21q的58.5%,其次为13q21q,占22%,而15q21q占19.5%。21qGq易位中,21q21q占83.3%,而21q22q仅占16.6%。无论体积易位,患者虽然只有46条体,但因一条21号易位到了另一条D组或G组染色体上,加上正常的两条21号,仍多了一条额外的21号长臂,而决定本病的关键区带为21号长臂,故临床上仍表现出21三体的症状。
(5)遗传学:典型的21三体几乎都是新发生(de novo)的,与父母的核型无关,经是减数分裂时不分离的结果。不分主离常发生在母方生殖细胞,约占病例数的95%,另5%见于父方,而且主要发生在第一次减数分裂。典型的21三体只有极少一部分是遗传的,即母亲是本病患者。此外,不能排除某些表型正常的母亲实际是21三体细胞较少的嵌合体,因而她们的子女有可能获得额外的21号染色体。男性患者不能生育,没有遗传给下一代的问题。
嵌合型患者有两个或两个以上的细胞系(图2-6)。它们是合子后(post-zygotic)有丝分裂不分离的结果。如果第一次卵裂时发生不分离,就会产生47,+21和45,-21两个细胞系。而后一种细胞是很维存活的。因此,导致嵌合体的不分离多半发生在以后的某次有丝分裂,所有嵌合体内都有正常的细胞系。
易位型的21三体征患者细胞中有一条易位的染色体,后者通常由一条D组或G组染色体与21号染色体长臂通过着丝粒融合(罗氏易位)而成。Dq21q易位中,55%是新发生的,45%是由于双亲之一有平衡易位。21qGq 易位几乎全部(96%)是新发生的,由遗传而来的仅占4%。
各种易位的遗传后果不同。Dq21q平衡易位的携带者通过减数分裂可以形成6种配子,而受精后除不能发育者外,可以产生正常胎儿、易位型三体患儿和平衡易位携带者三种胎儿(图2-12)。因此,检出平衡易位携带者的双亲具有重要意义。
21qGq易位中,21q22q和21q21q易位的遗传学意义不完全相同。如果双亲之一为21q21平衡易位携带者,就没有可能娩出表型正常的胎儿,因为他们只能产生三体或单体的合子。21q22q易位的遗传后果与Dq22q相似,只是前者更多通过父亲传递而后者多由母亲传递得来。
(6)遗传咨询:各种类型再发风险不同。对游离型的21三体性,再发风险率与年龄特异风险率相近(即35岁以下约为0.5%,35%岁以上约为1%)。然而,如双亲(通常是母亲)之一嵌合体则风险会增加。总之,已产生一个21三体患儿,其再发风险0.5%,而染色体畸变的总风险为1.2%,即不排除再生产其它染色体异常的患儿。
易位型有大约1/2的病例是新发生的,另1/2是双亲的之一平衡易位引起的。前者复发风险很小,而平衡易位导致的再发风险则可以根据经验估计。理论上如双亲之一为平衡易位携带者,产出患儿风险为33.3%。但实际风险低得多,这与双亲哪一方为携带者有关。Dq21q易位携带者若是母亲,生产患儿的风险为10%-15%;如为父亲,则风险为5%或更小。21q21q易位的情况与之大体相同,但易位染色体由父方传递的百分比较D/G易位要多,风险率在10%以下。21q21q平衡易位携带者的后代100%均是三体征患儿,携带者不宜生育。
(7)预后:患者平均寿命只有16.2岁。50%的患儿在5岁以前死亡。只有8%的患者超过40岁,2.6%超过50岁。根据四川省的资料,人群中患病率仅约为再生时的1/10。
2.13三体综合征1960年Patau首先描述本病,故又称为Patau综合征。新生儿中的发病率约为1:25 000,女性明显多于男性。
(1)临床表现:患儿的畸形和临床表现要比21三体性严重得多(图2-18)。颅面的畸形包括小头,前额、前脑发育缺陷,眼球小,常有虹膜缺损,鼻宽而扁平,2/3患儿有上唇裂,并常有腭裂,耳位低,耳廓畸形,颌小,其它常见多指(趾),手指相盖叠,足跟向后突出及足掌中凸,形成所谓摇椅底足。男性常有阴囊畸形和隐睾,女性则有阴蒂肥大,双阴道,双角子宫等。脑和内脏的畸形非常普遍,如无嗅脑,心室或心房间隔缺损、动脉导管未闭,多囊肾、肾盂积水等,由于内耳螺旋器缺损造成耳聋。
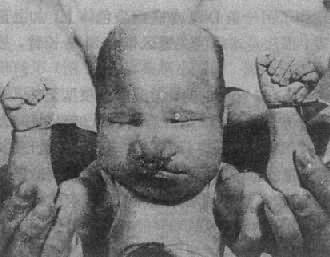
图2-18 Patau综合征患者示颅面和手的畸形
智力发育障碍见于所有的患者,而且程度严重,存活较久的患儿还有癫痫样发作,肌张功力低下等。
(2)细胞遗传学及遗传咨询:80%的病例为游离型13三体,核型为46,XX(或XY),+13,其余的则为嵌合型或易位型。嵌合型一般症状较轻,易位型通常以13和14号罗氏易位居多,患者有一条t(13q14q)易位染色体,核型为46,-14,+t(13q14q),其结果是多了一条13号长臂,当双亲之一是平衡易位携带者时,因为绝大多数异常胎儿均流产死亡,产出患儿的风险不超过5%或1%。如果双亲之一为13q13q易位携带者,由于只能产生三体或单体的合子,流产率达100%
(3)病因及预后:与13三 体发和表关的因素所知甚少,母亲高龄可能是原因之一,患儿母亲的平均年龄为31.6岁,父亲的平均年龄为34.6岁。此外,有资料表明,79%病例妊娠于寒冷季节(9-2月),45%的患儿在出生后一个月内死亡,90%在6个月内死亡,存活至3岁者少于5%,平均寿命为130天。
3.18三体综合征,1960年Edward等首先描述,故又称为Edward综合征(Edward syndrome ).18三体性导致严重畸形,在出生后不久死亡。发病率约1:3500-8000新生儿。但在某些地区或季节明显增高,达到1:450-800。患儿中女性:男性比为4:1。
(1)临床表现:患儿出生时体重低,平均仅2243g,发育如早产儿,吸吮差,反应弱,头面部和手足有严重畸形(图2-19),头长而枕部凸出,面圆,眼距宽,有内赘眦皮,眼球小,角膜混浊,鼻梁细长,嘴小,耳位低,耳廓畸形(动物样耳),小颌(micrognathia),颈短,有多余的皮肤,全身骨骼肌发育异常,胸骨短,骨盆狭窄,脐疝或腹股沟疝,腹直肌分离等。手的畸形非常典型:紧握拳,拇指横盖于其它指上,其它手指互相叠盖,指甲发育不全,手指弓形纹过多,约1/3患者为通贯掌。下肢最突出的是“摇椅底足”,拇趾短,向背侧屈起。外生殖器畸形比较常见的有隐睾或大阴唇和阴蒂发育不良等。95%的病例有先天性心脏病,如室间隔缺损、动脉导管未闭等,这是死亡的重要原因。肾畸形,肾盂积水也很常见。患儿智力有明显缺陷,但因存活时间很短,多数难以测量。
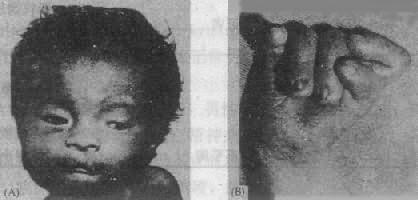
图2-19 18三体综合征患者(左)面部特征;(右)手的典型握拳式
(2)细胞遗传学:80%患者核型为47,XY(或XX),+18;另10%患者为嵌合体,即为46,XY(或XX)/47,XY(或XX),+18;其余为各种易位,主要是18号与D组染色体的易位。双亲是平衡易位携带者而导致18三体征者很少。
(3)预后;患儿大多在2-3个月内死亡,平均存活71天,只有极个别病人超过儿童期。嵌合型的存活期比较长。
4.其它染色体三体综合征比较重要的有8号、22号三体综合征等。都伴有明显的发育畸形和智力低下。还有一系列由易位引起染色体部分三体综合征,其临床症状取决于额外染色体片段的性质和大小。染色体部分三体性可分为两大类:一类有某一染色体片段的三体性(重复),同时又伴有其它染色体的异常(如缺失、易位),这一类部分三体性的表型比较复杂,常兼有重复和缺失片段的某些症状;另一类为染色体的某一片段的单纯重复或三体性,这在人类极为少见。
(二)单体及部分单体综合征
整条常染色体的丢失通常是致死的,因而极为罕见,但确有小染色体(如21号)完全丢失的报告。由于易位、环形成或缺失导致的染色体部分单体则比较多见。现以5q-综合征,即猫叫综合征为介绍如下。
猫叫综合征(5q-综合征)为最见的缺失综合征,其发病率估计为1:50000,女性多于男性。患婴的哭叫声非常似小猫的咪咪声,故得名。患儿面部情似很机灵,但实则智力低下非常严重(智商常低于20),发育迟滞也很明显。常见的临床表现还有小头、满月脸、眼裂过宽、内眦赘皮、下颌小且后缩(图2-20)。约20%患者有先天性心脏病,主要是室间隔缺损和动脉导管未闭等。
患者的染色体缺失片段大小不一。症状主要由5p15的缺失引起。畸变多数是新发生的。由染色体片段的单纯缺失(包括中间缺失)的占80%,不平衡易位引起的占10%,环状染色体或嵌全体则比较少见,由亲代染色体重排导致的5p-综合征不多见。
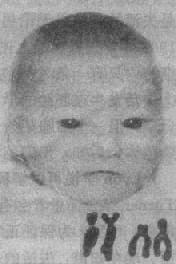
图2-20猫叫综合征患者及其部分核型
患者的死亡率低,许多能活到成年。
表2-6 亲代平衡易位携带者再出生不平衡易位患儿的概率(%)
| 易位类型 | 先证者 | 亲代携带者 | |
| 母 | 父 | ||
| 2平衡的相互易位 | 不平衡 | 15 7 | |
| 平衡的朴素易位 | 平衡 | 极微 | |
| D/G罗氏易位 | 不平衡 | 15 7 | |
| D/G罗氏易位 | 平衡 | 极微 | |
| D/G罗氏易位 | 平衡或不平衡 | 很小 | |
(引自Jacobs PA.1979)
四、性染色体异常综合征
(一)性别和性染色体
1.性别的决定人类有X和Y两种性染色体,但决定个体的表型性别是Y染色体。从临床上看,除个别例外,凡有Y者皆为男性,否则均为女性。因为Y染色体上有决定睾丸发育的睾丸决定因子(testis determining factor,TDF),而睾丸的存在决定了个体的性别。
2.X染色体的失活女性有两条X染色体而男性只有一条,但女性X染色体的基因产物并未比男性多1倍。这种男女X连锁基因产物相等的现象在遗传学一称为剂量补偿(dosage compensation)。对这一现象的解释是:虽然女性有两条X染色体,但其中一条是失活的,结果无论男女都只有一条有功能的X染色体。英国遗传学家Mary Lyon 在1961年首先提出了上述X染色体失活假说,即Lyon假说,其要点是:①雌性哺乳动物细胞内只有一条X染色体有活性,另一条失活并固缩,后者在间期细胞表现为性染色质;②失活发生在胚胎的早期;③失活是随机的,即失活的X染色体既可来自父亲也可来自母亲,但一个细胞某条X-旦失活,由该细胞繁衍而来的子细胞都具有同一条失活的X染色体。后知,X染色体失活发生在囊胚期,约在妊娠16天左右。
Lyon学说可以解释许多遗传现象,但经典的Lyon假说不能解释何以核型为XO的Turner综合征患者会有各种异常,又何以多X患者还会有各种症状,而且X越多症状越严重。可见,为保证正常的发育,至少在胚胎发育的某一时期需要双份X染色体上的基因。现在知道,失活的一条XX染色体上的基因并非全都失活,这方面已有一些证据。如已知Xg血型基因、寻常牛皮癣基因等是不失活的。有作者还提出,Y染色体有一些与X染色体型基因同源的基因,这样,正常男性或女性都有两份这类基因,但XO患者缺少一份而XXX患者有三份,因之都有表型异常。
失活的X染色质在间期呈固缩状态,称为X染色质(X-chromatin)。在口腔颊粘膜细胞或其它细胞中都可以用简单的染色方法查见(图2-21)。X染色质的数目是X染色体数目减1。这样,当怀疑有X染色体异常时,可以通过用性染色质检查作出初步诊断。例如,X染色质在XO时为0,正常女性为1,XXY患者为1,XXX患者为2。
(二)性染色体数目异常综合征
1.Klinefelter综合征(Klinefelter syndrome)又称为先天性睾丸发育不全或原发小睾丸症。患者性染色体为XXY,即比正常男性多了一条X染色体,因之本亦常称为XXY综合征。
Klinefelter综合征的发病率相当高,男性新生儿中达到1.2‰。根据白种人的资料,身高180cm的男性患病率为1/260,在精神病患者或刑事收容机构中为1/100,在因不育而就诊者中约为1/20。临床表现为睾丸小而质硬,曲细精管萎缩,呈玻璃样变。由于无精子产生,故97%患者不育。患者男性第二性征发育差,有女性化表现,如无胡须,体毛少,阴毛分布如女性,阴茎龟头小等,约25%的患者有乳房发户(图2-22)。患者身材高。四肢长,一部分患者(约1/4)有智力低下,一些患者还有精神异常及患精神分裂症倾向。实验室检查可见雌激素增多,19-黄体酮增高,激素的失调与患者的女性化可能有关。
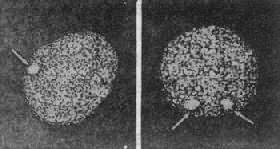
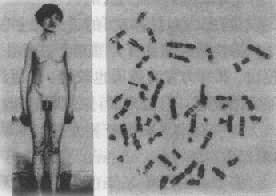
图2-21 X染色质 图2-22 Klinefelter综合征患者及其核型
绝大多数患者的核型为47,XXY。大约有15%患者为两个或更多细胞系的嵌合体,其中常见的为46,XY/47,XXY;46,XY/48,XXXY。额外的X是由于亲代减数分裂时X染色体不分离的结果。
用睾丸酮治疗可以收到一定的效果,它可促使第二性征发良并性病患者的心理状态。
2.XYY综合征在男婴中的发生率为1:900。XYY男性的表型的正常的,患者身材高大,常超过180cm,偶尔可见隐睾,睾丸发育不全并有精过程障碍和生育力下降,尿道下裂等,但大多数男性可以生育。XYY个体易于兴奋,易感到欲望不满足,厌学,自我克制力差,易产生攻击性行为。XYY核型是父亲精子形成过程中第二次减数分裂时发生Y染色体不分离的结果。
3.Turner综合征又称为45,X或45,XO综合征、女性先天性性腺发育不全或先天性卵巢发育不全综合征。在新生女婴中的发病率约为0.2‰-0.4‰,但在自发流产胚胎中Turner综合征的发生率可高达7.5%.患者表型为女性,身材矮小,智力一般正常,但常低于其同胞,面呈三角形,常有睑下垂及内眦赘皮等,上颌突窄,下颌小且后缩,口角下旋呈鲨鱼样嘴,颈部的发际很低,可一直伸延到肩部,约50%患者有蹼颈,即多余的翼状皮肤,双肩径宽,胸宽平如盾,乳头和乳腺发育差,两乳头距宽,肘外翻在本病十分典型(图2-23),第四、第五掌骨短而内

图2-23 Turner综合征患者及其核型
弯,并常有指甲发育不全。婴儿期脚背有淋巴样肿,十分特殊。泌尿生殖系统的异常主要是卵巢发育差(索状性腺),无滤泡形成,子宫发育不全,常因原发性闭经来就诊。由于卵巢功能低下患者的阴毛稀少,无腋毛,外生殖器幼稚。此外,大约有1/2患者有主动脉狭窄和马蹄肾等畸形。
Turner综合征的核型除典型的45,X(约占55%)外,还有各种嵌合型和结构异常的核型。最常见的是嵌合型46,XX/45,X和46,X,i(Xq)。一般说来,嵌合型的临床表现较轻,而有Y染色体的嵌合型可表现出男性化的特征,身材矮小和其它Turner症状主要是由X短臂单体性决定的,但卵巢发育不全与不育则更多与长臂单体性有关。
Turner综合征的发病机理是双亲配子形成过程中的不分离,其中约75%的染色体丢失发生在父方,约10%的丢失发生在合子后早期卵裂时。
除少数患者由于严重畸形有新生儿期死亡之外,一般均能存活,只是在青春期才被检出。其智力发育障碍也较轻,应用激素在14岁以前开始治疗可以促进第二性征和生殖器官的发育,月经来潮,心理状态改变,但不能促进长高,个别患者可生育。
4.47,XXX和X女性本病又称为超雌(superfemale)。发病率约为0.8‰或1/2250。
多数具有三条X染色体的女性无论外形、性功能与生育力都是正常的,只有少数患者有月经减少、继发闭经或过早绝经等现象。大约有2/3病智力稍低,并有患精神病倾向。
除了47,XXX外,一些患者的核型为嵌合型,即47,XXX/46,XX。XXX患者的母亲生育年龄平均约增高4岁。这表明不分离主要发生在母亲一方。少数患者有4条甚至5条X染色体。一般来说,X染色体愈多,智力损害和发育畸形愈严重。
(三)性染色体的结构畸变
1.X染色体的结构异常常见的X染色体结构异常有各种缺失、易位和等臂染色体。它们的临床表现多样,主要取决于涉及X染色体上的哪些区段异常,因为不同的区段载有的基因不同,缺失导致的症状也不同。Wyss等曾作一Turner综合征的核型与症状关系图。有助于了解X染色体结构异常的表现(图2-24)。
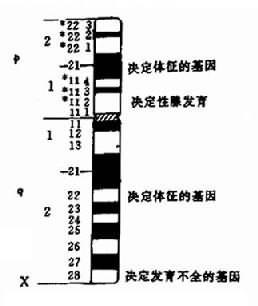
图2-24 Turner综合征核型与症状相关图
(1)X短臂缺失(XXp-):Xp远端缺失病人有诸如身材矮小等Turner综合征特征,但性腺功能正常。Xp缺失如包括整个短臂,则患者既有Turner综合征的体征,又有性腺发育不全。X染色体长臂等染色体[X,i,(Xq)]的临床表现与此类似,因为也缺失了整个短臂。
(2)X长臂缺失(XXq-):缺失在q22以远者,一般仅有性腺发育不全,原发闭经,不育,而无其它诸如身材矮小等Turner综合征体征。缺失范围较大,包括长臂近端者,有性腺发育不全外,一些患者还有其客观存在体征。X染色体等臂染色体[Xi(p)]与此类似。Xq中间缺失累及q13-q26者性腺功能正常,但有其它体征,可见中段缺失与Turner体征出现有关。
通常部分缺失`形成环状或等臂染色体的X均选择性地失省事,从而保证有一条正常的X。
(3)易位:当X染色体与常染色体发生平衡易位时,由于基因平衡的保持,一般不会产生症状,此时失活的正常的X染色体。但如平衡易位断点在q12-q26时,有活性的X在该区被分为两部分,就会导致性腺发育异常。此外,如常染色体节段易位到X染色体产生不平衡易位时,多数产生双着丝粒染色体,其表型取决于Xp或Xq上断裂点的位置。
2.脆性X染色体综合征本世纪初一些学者注意到智力下患者中逻辑性多于女性。1943年Martin和Bell在一个家系两代人中发现11名逻辑性患者和两名轻度智力低下的女性,认为该家系智力低下是与X连锁的,因此X连锁智力低下又称为Martin-Bell综合征。
1969年Lubs首先在逻辑性智力低下患者及其女性亲属中发现了长臂具有“随体和呈细丝状次缢痕”的X染色体。后来,Sortherland证明细丝位位于X染色体长臂2区7带(Xq27)。它在低叶酸培养条件下表达,并提出了脆性部位(franile site)的概念。现今人们把在Xq27处有脆性部位的X染色体称为X染色体(fragileX,fra X)(图2-25),而它所导致的疾病称为脆性X染色体综合征。

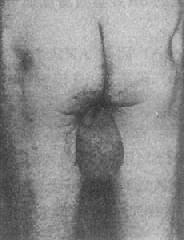
图2-25 脆性X染色体(Fra X)综合征
左图:脆性X染色体
中图:患者大耳`大颌
右图:患者大睾丸
(1)发病率:本病在逻辑性中的发病率为1/1000~1/1500,仅次于先天愚型。在所有逻辑性智力低下患者约10%~20~为本病所引起。
(2)临床表现:主要表现为中度到重度的智力低下,其它常见的特征尚有身长和体重超过正常儿,发育快,前额突出,面中部发育不全,下颌大而前突,大耳,高腭弓,唇厚,下唇突出,另一个重要的表现是大睾丸症。一些患者还有多动症,攻击性行为或孤癖症。20%患者有癫痫发作。过去曾认为由于女性有两条X染色体,因此女性携带者不会发病,但由于两条X染色体中有一条失活,女性杂合子中约1/3可有轻度智力低下。
(3)发病的分子机理:现今在X脆性部们已发现了致病基因FMR-1,它含有(CGC)n 三核甘酸重复序列,后者在正常人约为30拷贝,而在正常男性传递者和女性携带者增多到150~500bp,称为小插入,相邻的Cpg 岛未被甲基化,这种前突变(premutation )无或只有轻微症状。女性携带者的CGG区不稳定,在向受累后代传递过程中扩增,以致在男性患者和脆性部位高表达的女性达到1000~3000bp,相邻的CpG岛也被甲基化。这种全突变(full mutation)可关闭相邻基因的表达,从而出现临床症状。由前突变转化为完全突变只发生母亲向后代传递过程中。根据对脆性部位DNA序列的了解,现已可用RFLP连锁分析、DNA杂交分析、PCR扩增等方法来检出致病基因。
(4)治疗:Lejeune 认为叶酸缺乏是Fra X综合征时智力低下的原因,他用大剂量叶酸治疗患者获得了良好的效果,但其他作者未能证实叶酸的疗效。新近一些作者认为中枢神经兴奋剂疗效较好,但副作用大。其它有用可乐定(clonidine)、心得安者,据称可减轻多动症。
3.Y染色体及其结构异常 Y染色体用荧光染色时,因长臂未端有宽阔的极明亮的荧光带,易与21、22号区别。Y染色体长臂的多态性非常明显,存在种族差异,大Y在中国人和日本人中的比例较高。
4.Y染色体的数目异常 包括前已述及的XYY以及超Y综合征(XXYY,X--等)。Y染色体的结构异常包括Y的长臂或短臂缺失、等臂染色体i(Yq)和i(Yp)、环状染色体和双着丝粒染色体(为两条Y的短臂 相连或两条Y的长臂相融合)、倒位和各种涉及Y的易位(即Y与常染色体,Y与X染色体的易位等)。此外性反转综合征46,XX男性是Yq上的SRY基因易位到一条X染色体所致,而46,XY女性是SRY基因缺失或突变的结果。
第三章 基因及基因突变
第一节 基因的概念
基因的概念随着遗传学、分子生物学、生物化学等领域的发展而不断完善。从遗传学的角度看,基因是生物的遗传物质,是遗传的基本单位——突变单位、重组单位和功能单位;从分子生物学的角度看,基因是负载特定遗传信息的DNA分子片段,在一定条件下能够表达这种遗传信息,变成特定的生理功能。有的生物基因为RNA。
一、基因的一般的特性
从分子水平来说,基因有三个基本特性:①基因可自体复制;②基因决定性状,即基因通过转录和翻译决定多肽链的氨基酸顺序,从而决定某种酶或蛋白质的性质,而最终表达为某一性状;③基因的突变,即基因虽很稳定,但也会发生突变。一般来说,新的突变的等位基因一旦形成,就可通过自体复制,在随后的细胞分裂中保留下来。
二、基因的类别
基因按其功能可分为:
1.结构基因(structural gene)是指某些能决定某种多肽链(蛋白质)或酶分子结构的基因。结构基因的突变可导致特定蛋白质(或酶)一级结构的改变或影响蛋白质(或酶)量的改变。
2。调控基因(regulator and control gene)是指某些可调节控制结构基因表达的基因。调控基因的突变可以影响一个或多个结构基因的功能,或导致一个或多个蛋白质(或酶)时的改变。
此外,还有一些只转录而不翻译的基因,如核糖体RNA基因(ribosomal RNA gene),也称为rDNA基因,它们专门转录rRNA;还不转运RNA基因(transfer RNA gene),也称为tRNA基因,是专门转录tRNA的。
存在于原核生物与真核生物中的基因也有区别:
1.原核生物 一般只有一个染色体,即一个核酸分子(DNA或RNA),大多数为双螺旋结构,少数以单链形式存在。这些核酸分子大多数为环状,少数为线状。例如大肠杆菌染色体是由4.2×106bp(碱基对)组成的双链环状DNA分子,约有3000~4000个基因,目前已经定位的基因已达900多个。
2.真核生物包括人类在内,其基因主要存在于细胞核内线状的染色体上。存在于细胞质的基因位于环状的线粒体DNA上。核内基因的DNA顺序由编码顺序和非编码顺序两部分构成。编码顺序是不连续的,被非编码顺序隔开。其次,真核的生物基因大小差别很大,例如,人类血红蛋白的基因长仅约1700bp,而假肥大型营养不良症(duchenne muscular dystrophy, DMD)基因全长2300kb,是迄今认识的最巨大的人类基因。
第二节 结构基因的结构
人类结构基因4个区域:①编码区,包括外显子与内含子;②前导区,位于编码区上游,相当于RNA5’末端非编码区(非翻译区);③尾部区,位于RNA3’编码区下游,相当于末端非编码区(非翻译区);④调控区,包括启动子和增强子等。基因编码区的两侧也称为侧翼顺序(图3-1)。
1.外显子和内含子 大多数真核生物的基因为不连续基因(interruptesd或discontinuous gene)。所谓不连续基因就是基因的编码顺序在DNA分子上是不连续的,被非编码顺序所隔开。编码的顺序称为外显子(exon),是一个基因表达为多肽链的部分;非编码顺序所称为内含子(intron),又称插入顺序(intervening sequence,IVS)。内含子只转录,在前mRNA(pre-mNRA)时被剪切掉。如果一个基因有几个内含子,一般总是把基因的外显子分隔成n+1部分。内含子的核苷酸数量可比外显子多许多倍。
2.外显子-内含子 接头每个外显子和内含子接头区都有一段高度保守的一致顺序(consensus seqence),即内含了5’末端大多数是GT开始,3’末端大多是AG结束,称为GT-AG法则,是普遍存在于真核基因中RNA剪接的识别信号。
3.侧翼顺序在第一个外显子和最末一个外显子的外侧是一段不被翻译的非编码区,称为侧翼顺序(flankingsequence)。侧翼顺序含有基因调控顺序,对该基因的活性有重要影响。
4.启动子 启动子(promoter)包括下列几种不同顺序,能促进转录过程:
(1)TATA框(TATA box):其一致顺序为TATAATAAT。它约在基因转录起始点上游约-30-50bp处,基本上由A-T碱基对组成,是决定基因转录始的选择,为RNA聚合酶的结合处之一,RNA聚合酶与TATA框牢固结合之后才能开始转录。]
(2)CAAT框(CAAT box):其一致顺序为GGGTCAATCT,是真核生物基因常有的调节区,位于转录起始点上游约-80-100bp处,可能也是RNA聚合酶的一个结合处,控制着转录起始的频率。
(3)GC框(GC box):有两个拷贝,位于CAAT框的两侧,由GGCGGG组成,是一个转录调节区,有激活转录的功能。
此外,RNA聚合酶Ⅲ负责转录tRNA的DNA和5SrDNA,其启动子位于转录的DNA顺序中,称为下游启动子。
5.增强子在真核基因转录起始点的上游或下游,一般都有增强子(enhancer),它不能启动一个基因的转录,但有增强转录的作用。此外,增强子顺序可与特异性细胞因子结合而促进转录的进行。研究表明,增强子通常有组织特异性,这是因为不同细胞核有不同的特异因子与增强子结合,从而对基因表达有组织、器官、时间不同的调节作用。
例如人类单拷贝胰岛素基因5’末端上游约250 bp处有一组织特异性增强子,在胰岛β细胞中有一特异因子可
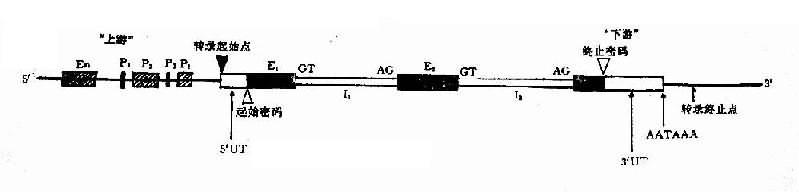
图3-1 真核生物的结构基因的结构示意图
En:增强子:P1、P2、P3:启动子(TATA框,CAAT框,GC框);E:外显子:I:内含子;
UT:非翻译区;GT-AG:外显子-内含子接头
作用于该区以增强胰岛素基因的转录和翻译,其它组织中无此因子,这是何以胰岛素基因只有在胰岛β细胞中才得以很好表达的原因。
6.终止子在一个基因的末端往往有一段特定顺序,它具有转录终止的功能,这段终止信号的顺序称为终止子(termianator)。终止子的共同顺序特征是在转录终止点之前有一段回文顺序,约7-20核苷酸对。回文顺序的两个重复部分分由几个不重复碱基对的不重复节段隔开,回文顺序的对称轴一般距转录终止点16-24bp(图3-2)。
在回文顺序的下游有6-8个A-T对,因此,这段终止子转录后形成的RNA具有发夹结构,并具有与A互补的一串U,因为A-U之间氢健结合较弱,因而RNA/DNA杂交部分易于拆开,这样对转录物从DNA模板上释放出来是有利的,也可使RNA聚合酶从DNA上解离下来,实现转录的终止。
图3-2 转录终止了顺序图解
第三节 基因的复制与表达
生物的遗传物质基础是核酸(nucleicacid),它也是基因的基本结构,它们的化学组成分子结构符合遗传物质的稳定性、连续性及多样性的要求。
(一)核酸的化学组成
核酸结构的基本单位是核苷酸(nucleicacid),每个核苷酸由1个磷酸、1个五碳糖和1个碱基3部分组成。核酸有两类:一类是脱氧核糖核酸(DNA),DNA中的脱氧核糖核苷酸主要由4种碱基构成,即腺嘌呤(adenine,A)、鸟嘌呤(guanine,G)、胞嘧啶(cytosine,C)和胸腺嘧啶(thymine,T),此外,还有脱氧核糖(deoxyribose)和磷酸;另一类是核糖核酸(RNA),RNA分子中的核糖核苷酸主要由碱基A、G、C和尿嘧啶(uracil,U)构成,此外,还有核糖(ribose)和磷酸。
(二)DNA的分子结构
DNA分子是4种脱氧核苷酸经3’→5’磷酸二酯健聚合而成,所以了称为多核苷酸(polynucleotide)。DNA的一级结构是指4种核苷酸的连接及其排列顺序。1953年Watson和Cricd提出了DNA分子双螺旋结构模型,其要点是:DNA分子是由2条平行的多核苷酸链围绕同一中心轴构成的右手双螺旋结构(B型DNA)。多种芏酸的方向由核苷酸间的磷酸二酯健的走向决定,一条从5’→3’,另一条从3’→5’,两条链呈反向平行排列(antiparallel),彼此由氢键相连,G 与C配对(G≡C),A与T配对(A=T)。图3-3表明DNA的分子骨架。
图3-3 DNA双螺旋结构及碱基配对示意图
(A)部分SNA多核苷酸链,示邻近脱氧核苷酸由3’-5’磷酸二酯键连接;
(B)DNA互补的两条链;(C)DNA双螺旋模型
根据以上原则,只要确定了一条链中的碱基顺序,就可以相应在确定与它互补的另一条链上碱基的顺序,估计1个DNA分子大约有4千至40亿个核酸对,而各种碱基对排列顺序没有限制,即假定某一段DNA分子链有100个碱基对,则该段就有4100各不同的排列组合形式,即可有4100种不同性质的基因。现知,基因就是DNA分子链上的一个特定的区段,其平均大小约为1 000个碱基对。这说明对DNA分子贮存了大量正常和变异的遗传信息,满足了生物的遗传和多样性的要求,特别是通过DNA分子的准确复制,又可使遗传信息得到稳定和连续的传递。
(三)DNA的复制
基因的复制的以DNA复制为基础的。生物体的遗传特征表现为特定的核苷酸顺序,并以密码子的形式编码在DNA分子上。在细胞分裂过程中,通过DNA准确地自体复制(self-replication),把遗传信息从亲代传给子代,这样,DNA就能真正完成其作为遗传信息载体的使命,从而保证遗传物质的连续性和相对的稳定性。
由于DNA分子两条链的碱基是互补的,一条链上的核苷酸排列顺序可以由另一条链上的核苷酸排列顺序决定。DNA复制过程中,首先碱基间氢断裂,双螺旋解旋并松开,然后每条多核苷酸链各以自己为模板(template)吸收周围游离核苷酸,按碱基互补原则,进行氢键结合。在一些聚合酶作用下,合成新的互补的链,与原来模板单链并列盘旋在一起,形成了稳定的双螺旋结构(图3-4)。
这样新形成的2个DNA分子与原来 DNA分子的碱基顺序完全一样。每个子代DNA分子的一条链来自亲代DNA,另一条链则是新合成的,所以这种复制方式称为半保留复制(swmi-conservativereplication).
图3-4 双螺旋DNA半保留复制过程
P1,P2:DNA亲链;F1,F2:DNA子链;
示复制结果的两个DNA分子完全相同,每个新DNA分子均由亲链和子链组成
(四)基因的表达
所谓基因表达(geneexpression)是指细胞在生命过程中,把储存在DNA顺序中遗传信息经过转录和翻译,转变成具有生物活性的蛋白质分子.生物体内的各种功能蛋白质和酶都是同相应的结构基因编码的(图3-5)。
1.转录过程在RNA聚合酶的催化下,以DNA为模板合成mRNA的过程称为转录(transcription).在双链DNA中,作为转录模板的链称为模板链(template strand),或反义链(antisensestrand);而不作为转录模板的链称为编码链(codingstrand),或有义链(sense strand).在双链DNA中与转录模板互补的一条DNA链即编码链,它与转录产物的差异仅在于DNA中T变为RNA中的U.在含许多基因的DNA双链中,每个基因的模板链并不总是在同一条链上,亦即一条链可作为某些基因的模板链的,也可是另外一些基因的编码链。
转录后要进行加工,转录后的加工包括:
(1)剪接:一个基因的外显子和内含子都转录在一条原始转录物RNA分子中,称为前mRNA(pre-mRNA),又称核内异质RNA(heterogenuous nuclear RNA,huRNA)。因此前mRNA分子既有外显子顺序又有内含子顺序,另外还包括编码区前面及后面非翻译顺序。这些内含子顺序必须除支而把外显子顺序连接起来,才能产生成熟的有功能的mRNA分子,这个过程称为RNA剪接(RNasplicing)。剪切发生在外显子的3’末端的GT和内含子3’ 末端与下一个外显子交界的AG处。
(2)加帽:几乎全部的真核 mRNa 端都具“帽子”结构。虽然真核生物的mRNA的转录以嘌呤核苷酸三磷酸(pppAG或pppG)领头,但在5’端的一个核苷酸总是7-甲基鸟核苷三磷酸(m7GpppAGpNp)。mNRA5’端的这种结构称为帽子(cap)。不同真核生物的mRNA具有不同的帽子。
mRNA的帽结构功能:①能被核糖体小亚基识别,促使mRNA和核糖体的结合;②m7Gppp结构能有效地封闭RNa 5’末端,以保护mRNA免疫5’核酸外切酶的降解,增强mRNA的稳定
(3)加尾:大多数真核生物的mRNA 3’末端都有由100~200个A组成的Poly(A)尾巴。Poly(A)尾不是由DNA编码的,而是转录后的前mRNA以ATP为前体,由RNA末端腺苷酸转移酶,即Ploy(A)聚合酶催化聚合到3’末端。加尾并非加在转录终止的3’末端,而是在转录产物的3’末端,由一个特异性酶识别切点上游方向13~20碱基的加尾识别信号AAUAAA以及切点下游的保守顺序GUGUGUG,把切点下游的一段切除,然后再由Poly(A)聚合酶催化,加上Poly(A)尾巴,如果这一识别信号发生突变,则切除作用和多聚腺苷酸化作用均显著降低(图3-5)。
图3-5 真核生物结构基因表达(DNA→RNA→蛋白质)流程图
mRNAPoly(A)尾的功能是:①可能有助mRNA从核到细胞质转运;②避免在细胞中受到核酶降解,增强mRNA的稳定性。
2.翻译过程真核细胞的转录以及加工都是细胞核内进行,但翻译过程则在细胞质中进行。
以mRNA作为模板,tRNA作为运载工具,在有关酶、辅助因子和能量的作用下将活化的氨基酸在核糖体(亦称核蛋白体)上装配为蛋白质多肽链的过程,称为翻译(translation),这一过程大致可分为3个阶段 (图3-6):
(1)肽链的起始:在许多起始因子的作用下,首先是核糖体的小亚基和mRNA上的起始密码子结合,然后甲酰甲硫氨酰tRNA(tRNA fMet)结合上去,构成起始复合物。通过tRNA的反密码子UAC,识别mRNA上的起始密码子AUG,并相互配对,随后核糖体大亚基结合到小亚基上去,形成稳定的复合体,从而完成了起始的作用。]
(2)肽链的延和长:核糖体上有两个结合点——P位和A位,可以同时结合两个氨酰tRNA。当核糖体沿着mRNA从5’→3’移动时,便依次读出密码子。首先是tRNAfMet结合在P位,随后第二个氨酰tRNA进入A位。此时,在肽基转移酶的催化下,P位和A位上的2个氨基酸之间形成肽键。第一个tRNA失去了所携带的氨基酸而从P位脱落,P位空载。A位上的氨酰tRNA在移
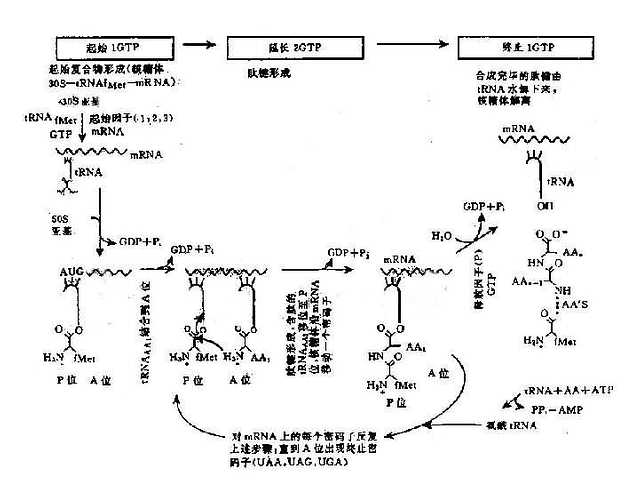
图3-6 翻译过程(蛋白质合成)图解
位酶和GTP的作用下,移到P位,A位则空载。核糖体沿mRNA 5’端向3’端移动一个密码子的距离。第三个氨酰tRNA进入A位,与P位上氨基酸再形成肽键,并接受P位上的肽链,P位上tRNA释放,A位上肽链又移到P位,如此反复进行,肽链不断延长,直到mRNA的终止密码出现时,没有一个氨酰tRNA可与它结合,于是肽链延长终止。
(3)肽链的终止:终止信号是mRNA上的终止密码子(UAA、UAG或UGA)。当核糖体沿着mRNA移动时,多肽链不断延长,到A位上出现终止信号后,就不再有任何氨酰tRNA接上去,多肽链的合成就进入终止阶段。在释放因子的作用下,肽酰tRNA的的酯键分开,于是完整的多肽链和核糖体的大亚基便释放出来,然后小亚基也脱离mRNA.
(4)翻译后加工(postranslational processing):从核糖体上释放出来的多肽需要进一步加工修饰才能形成具有生物活性的蛋白质。翻译后的肽链加工包括肽链切断,某些氨基酸的羟基化、磷酸化、乙酰化、糖基化等。真核生物在新生手肽链翻译后将甲硫氨酸裂解掉。有一类基因的翻译产物前体含有多种氨基酸顺序,可以切断为不同的蛋白质或肽,称为多蛋白质(polyprotein)。例如胰岛素(insulin)是先合成86个氨基酸的初级翻译产物,称为胰岛素原(proinsulin),胰岛素原包括A、B、C三段,经过加工,切去其中无活性的C肽段,并在A肽和B肽之间形成二硫键,这样才得到由51个氨基酸组成的有活性的胰岛素。
3.外显子与内含子表达过程中的相对性从内含子与外显子的定义来看,两者是不能混淆的,但是真核生物的外显子也并非都“显”(编码氨基酸),除了tRNA基因和rRNA基因的外显子完全“不显”之外,几乎全部的结构基因的首尾两外显子都只有部分核苷酸顺序编码氨基酸,还有完全不编码基酸的外显子,如人类G6PD基因的第一外显子核苷酸顺序。
现在已发现一个基因的外显子可以是另一基因的内含子,所这亦然。以小鼠的淀粉酶基因为例,来源于肝的与来源于唾液腺的是同一基因。淀粉酶基因包括4个外显子,肝生成的淀粉酶不保留外显子1,而唾液腺中的淀粉酶则保留了外显子1的50bp顺序,但把外显子2与前后两段内含子一起剪切掉,经过这样剪接,外显子2就变成唾液淀粉酶基因中的内含子。
4.同一基因在不同组织能生成不同的基因产物来源于不同组织的类似蛋白,可以由同一基因编码产生,这种现象首先是由于基因中的增强子等有组织特异性,它能与不同组织中的组织特异因子结合,故在不同组织中同一基因会产生不同的转录物与转录后加工作用。此外真核生物基因可有一个以一的poly(A)位点,因此能在不同的细胞中产生具有不同3’末端的前mRNA,从而会有不同的剪接方式。由于大多数真核生物基因的转录物是先加poly(A)尾巴,然后再行剪接,因此不同组织、细胞中会有不同的因子干预多聚腺苷酸化作用,最后影响剪接模式。降钙素(calcitonin)基因在不同组织中的表达可作为实例(图3-7)。
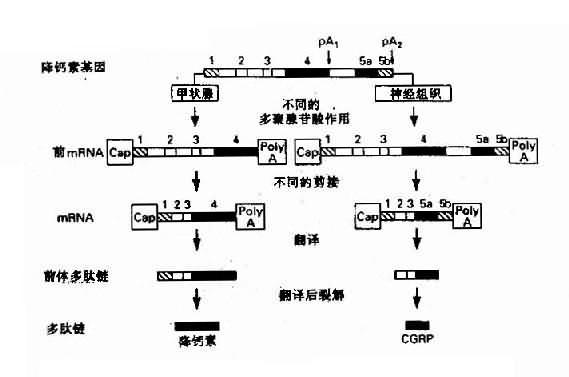
图3-7 降钙基因在不同的细胞中产生不同的激素
1:为非翻译顺序;2、3:为共同的编码外显子;
4:为降钙素外显子;5 a:为CGRP外显子;5b:为CGRP3’ 非翻译顺序;pA1、pA2:为AATAAA加尾信号;CGRP:与钙素基因相关的多肽
降钙素基因(CALCAa多肽,定位于11p15.4)中甲状腺细胞中形成的前mRNA(短转录物),包含有非翻译顺序(1)、编码外显子(2)和(3)以及降钙素编码外显子(4)(包括部分非编码区),在转录物pA1位点即AAUAAA信号附近进行多聚腺苷酸化。而在下丘脑,其前mRNA(长转录物)中除了包含转录物的全部顺序外,还包含有与降钙素基因相关肽(calcitonin gene-related peptide, CGRP)的编码外显子(5a)和CGRP的3’末端非翻译顺序(5b),并在转录物pA2处进行poly(A)加工。可是,在长转录物的剪接过程中,外显子(3)的拼接点直接与CGRP编码外显子(5a)拼接点相连,从而删除降钙素的编码外显子(4),这样形成两种成熟的mRNA,分别翻译产生降钙素的前体和CGRP前体,然后通过酶促降产生降钙素CGRP这两种激素。
第四节 人类基因组
人类基因组包括细胞核内的基因组及细胞质内线粒体基因组,它们大致结构如图3-8。
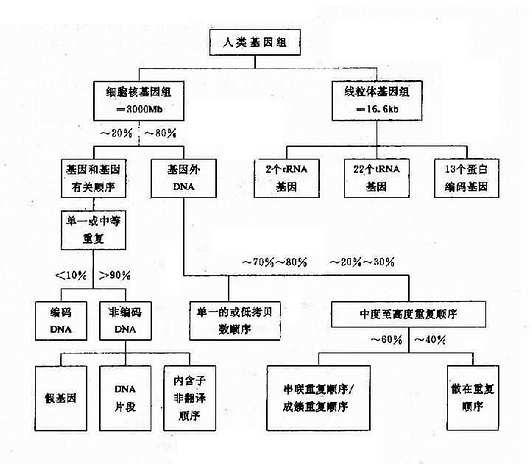
图3-8 人类基因组结构
一、细胞核基因组
每条染色体含1个DNA分子,1个细胞的全部遗传信息(基因)都编码在线状的DNA分子上。由于每个体细胞中有2套染色体(2n),故所含的DNA是由两个基因组(genome)构成。每个单倍体基因组约含3.2×109bp。人类基因的平均长度为1-1.5kb,所以基因组以足以编码1.5×106蛋白质,但实际上编码蛋白质的结构基因只不过5万-10万个,仅占总基因组的2%-3%。其余的DNA顺序包括基因之间的间隔顺序、基因内插入顺序、重复顺序等。目前,对它们的功能知之甚少,绝大多数重复顺序只不过是过剩的DNA。但是,其中一些则有着特殊的功能,包括:调节基因的表达,增强同源染色体之间的配对和重组,维持染色体结构,调节前mRNA的加工以及参与DNA的复制等。
(一)单一顺序
单一顺序(uniquesequence)约占基因组的60%-65%,这种顺序在一个基因组中一般仅有单个或几个拷贝,大多数编码蛋白质和酶基因属于此类。单一顺序还以间隔顺序和散在分布在重复顺序构成侧翼。
(二)重复顺序
重复顺序(repetitivesequence)是指在一个基因组中有很多拷贝,又可分为几类:
1.高度重复顺序(highly repetitive sequence) 其长度可能2、4、6、8等几个bp,较长的顺序可达200bp,但是重复拷贝数可达106次以上,例如染色体着丝粒、端粒和Y染色体长臂上的异染区就是由高度重复顺序的卫星DNA(satellite DNA)构成的,高度重复顺序不能转录,它们参与染色体结构的维持,形成结构基因间隔,可能与减数分裂时同源染色体的联会配对有关。
2.中度重复顺序(moderately repetitive sequence) 其长度300-7000bp,一般都是不编码的顺序。据认为在基因调控中起重要作用,包括开启或关闭基因,促进或终止转录,DNA复制的起始,参与前mRNA加工等。例如人类Alu家族(Alu family),占人类基因组的3%-6%,由300bp构成,在第170位附近都AGCT顺序,可被内切酶AluⅠ所切割(AG↓CT)故得名。这些顺序在基因中重复达30-50万次,平均5kbDNA就有一个Alu顺序。此外还有KpnⅠ家族(KpnⅠfamily),约占基因组的3%-6% ,由3000-4800个拷贝构成,其功能不详。此外,还有小卫星DNA和微卫星DNA(参阅第十三章)。
3.基因家族和基因簇真核基因组中有许多来源相同.结构相似.功能相关的基因,这组基因称为基因家族(genefamily)。基因家族的成员可以分布于几条不同染色体上,也可集中于一条染色体上。集中成簇的一组基因称为基因簇(gene cluster)。例如人类白细胞抗原(HLA)系统的7个连锁基因座位,排列成A-C-B-D-DR-DQ-DP,形成一个基因簇。此外,人类的类α和β珠蛋白基因簇分别集群串联排列于16p13和11p15上,而组蛋白基因簇则群集于7q32-q36。有些基因家族的成员并不集中排列为基因簇,而是散布在基因组中不同部位,如微管蛋白基因家簇,微管相关蛋白2(MAP2)定位于2q34-q35,微管相关蛋白tau,β(MAPT1)定位于17q21,微管相关蛋白tau-2(MAPT2)定位于6q21。
(三)假基因
在基因家族中的某些成员并不产生有功能的基因产物,称为假基因(pseudogene),如Ψξ、Ψα、Ψβ等。假基因起始也可能有功能,后来由于缺失、倒位或点突变等原因使这些基因成为无功能的基因。假基因可以与有功能基因连锁,也可以由于染色体易位或作为转座子,从一部位移到另一新的部位。
此外,人类基因组中还有一些特殊的短顺序位于各基因的侧翼,它们是起到调控作用的调节顺序(启动子、增强子等)。有的是与细胞恶性转化有关的原癌基因(proto-onco-gene)等构成了五花八门的人类基因组结构。
二、线粒体基因组
人类线粒体DNA(mitochondrial DNA,mtDNA)是独立于细胞核染色体外的又一基因组,它能自主复制,由16569个碱基对组成,每一个mtDNA分子为环状双链DNA分子,外环为重链,内环为轻链。基因组含有37个基因,其中13个为蛋白质基因(包含1个细胞素b基因,2个ATP酶复合体组成成分基因,3个细胞色素c氧化酶亚单位的基因及7个呼吸链NADH脱氢酶亚单位的基因),2个为rRNA基因,还有22个tNRA基因(图3-9)。
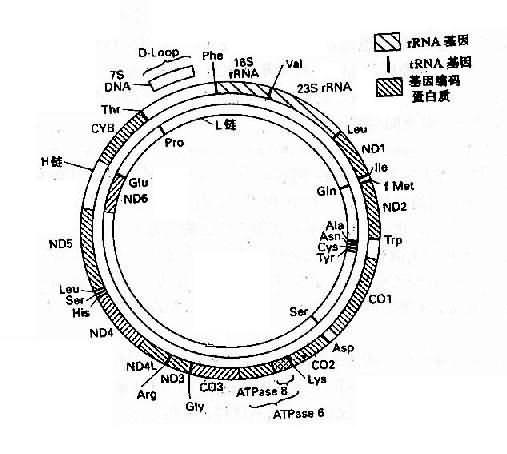
图3-9 人类线粒体基因组 H:重链;L:轻链
ND1-ND6:基因编码NADH脱氢酶亚单位CO1-CO3:基因
编码细胞色素C氧化酶亚单位1-3CYB:基因编码细胞色素b
人类线粒体基因组具有下列特点:
1.人类线粒体的基因排列得非常紧凑,除与mtDNA复制及转录有关的一小段区域外,无内含子序列。在37个基因之间,基因间隔区总共只有87bp,只占DNA总长度的的0.5%,有些基因之间没有间隔,有时基因有重叠,即前一个基因的最后一段碱基与下一个基因的第一段碱基相衔接。因此,mtDNA的任何突变都会累及到基因组中一个重要功能区域。
2.mtDNA为高效利用DNA。有5个阅读框架,缺少终止密码子,仅以U或UA结尾。
3.mtDNA的突变率高于核中DNA,并且缺乏修复能力。
4.mtDNA为母系遗传。
5.部分mtDNA的密码子不同于核内DNA的密码子。
遗传密码是在长期进化中形成并保持不变的,因此细胞核内所列的密码是一种通用密码,但是真核生物线粒体的密码却有若干处不同于通用密码。以人类线粒为例:①UGA不是终止密码子,而是色氨酸的密码子;②AGA、AGG不是精氨酸的密码子而是终止密码子。这样,加上通用密码中的UAA和UAG,线粒体共有4个终止密码子;③内部甲硫氨酸密码子有2个,即AUG和AUA,起始甲硫酸密码子有4个,即AUN。
总之,线粒体的密码表排列得比较整齐,各种氨基酸的密码子以及起始和终止密码子的数目或是2,或是4,或是6。
目前,由于发现有些遗传病如Leber遗传性视神经病,肌阵挛性癫痫等与线粒体基因突变有关,因而它的基因结构引起普遍关注。
第五节 基因突变
一、基因突变的特性
1.突变突变(mutation)是指遗传物质发生的可遗传的变异。广义的突变可以分两类:①染色体畸变(chromosome aberration),即染色体数目和结构的改变;②基因突变(genemutation)。狭义的突变,即一般所指的突变仅指基因突变。基因突变是指基因的核苷酸顺序或数目发生改变。仅涉及DNA分子中单个碱基改变者称点突变(point mutation)。涉及多个碱基的还有缺失、重复和插入。
2.体细胞突变和生殖细胞突变基因突变可发生在个体发育的任何阶段,以及体细胞或生殖细胞周期的任何分期。如果突变发生在体细胞中,突变的变异只能在体细胞中传递。因此体细胞突变不能直接遗传下代。生殖细胞的突变率比体细胞高,主要因为生殖细胞在减数分裂时对外界环境具有较高的敏感性。如果显性突变基因在生殖细胞中发生,它们的效应可能通过受精卵而直接遗传后代并立即在子代中表现出来;如果突变基因是隐性的,则其效应就可能被其等位基因所遮盖。如果突变发生在某一配子中,那么,在子代中只有某一个有可能承继这个突变基因。如果突变发生在配子发生的早期阶段(如发生在成熟分裂的性母细胞),则多个配子都有可能接受这个突变基因,因此,突变基因传到后代的可能性就会增加。携带突变基因的细胞或个体,称为突变体(mutant)没有发生基因突变的细胞或个体称为野生型(wild type)。
3.诱发突变和自发突变引起突变的物理因素(如X射线)和化学因素(如亚硝酸盐)称为诱变剂(mutagen)。诱变剂诱发的突变称为诱发突变(induced mutation)。由于自然界中诱变剂的作用或由于偶然的自制、转录、修复时的碱基配对错误所产生的突变称为自发突变(spontaneous mutation)。人类单基因病大都为自发突变的结果。自发突变频率(突变率)很低,平均每一核苷酸每一世代为10-10-10-9,即每世代10亿个核苷酸有一次突变。
4.突变热点从理论上讲,DNA分子上每一个碱基都可能发生突变,但实际上突变部位并非完全随机分布。DNA分子上的各个部分有着不同的突变频率,即DNA分子某些部位的突变频率大大高于平均数,这些部位就称为突变热点(hot spots of mutation)。形成突变热点的原因仍未明了,有人认为是5-甲基胞嘧啶(MeC)的存在,MeC脱氨氧化后生成T,引起G-MeC→A-T转换;短的连续重复顺序处容易发生插入或缺失突变;有的与突变剂种类有关,如DNA顺序中某个碱基对突变剂更敏感,有的则相反。
二、基因突变的种类
从DNA碱基顺序改变来分,突变一般可分为碱基置换突变、移码突变、整码突变及染色体错误配对和不等交换4种。
(一)碱基置换突变
一个碱基被另一碱基取代而造成的突变称为碱基置换突变(图3-10)。凡是一个嘌呤被另
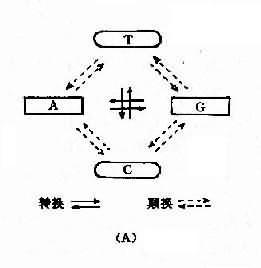
图3-10碱基置换类型(A)及缺失和插入突变(B)示意图
一个嘌呤所取代,或者一个嘧啶被另一个嘧啶所取代的置换称为转换(transition);一个嘌噙被另一个嘧啶所取代或一个嘧啶被另一个嘌呤所替代的置换称为颠换(transversion)。由此可产生4种不同的转换和8种不同的颠换。但自然界的突变,转换多于颠换。碱基置换会导致蛋白一级结构氨基酸组成的改变而影响蛋白质酶生物的功能。
由于碱基置换导致核苷酸顺序的改变,对多肽链中氨基酸顺序的影响,有下列几种类型;
1.同义突变由于密码子具有兼并性,因此,单个碱基置换后使mRNA上改变后的密码子与改变前所编码的氨基酸一样,肽链中出现同一氨基酸。例如DNA分子模板链中GCG的第三位G被A取代而成GCA,则mRNA中相应的密码子CGC就被转录为CGU,CGC和CGU都是精氨酸的密码子,翻译成的多肽链没有变化,这种突变称为同义突变(same-sense or synonymous mutation)。同义突变不易检出。据估计,自然界中这样的突变频度占相当高比例。
2.错义突变是指DNA分子中的核苷酸置换后改变了mRNA上遗传密码,从而导致合成的多肽链中一个氨基酸被另一氨基酸所取代,这种情况称为错义突变(missense mutation)。此时,在该氨基酸前后的氨基酸不改变。例如mRNA分子正常编码顺序为:UAU(酪)GCC(丙)AAA(赖)UUG(亮)AAA(赖)CCA(脯),当第三密码子A颠换为C时,则AAA(赖)→ACA(苏),即上述顺序改变为UAU(酪)GCC(丙)ACA(苏)UUG(亮)AAA(赖)CCA(脯)。错义突变结果产生异常蛋白质和酶。但也有不少基因由于错义突变而产生部分降低活性和异质组分的酶,从而不完全抑制了催化反应,这种基因称为漏出基因(leaky gene)。如果由于基因错义突变置换了酶活性中心的氨基酸,因此合成了没有活性的酶蛋白,虽不具有酶活性但有时还具有蛋白质抗原性,其所产生的抗体可与正常蛋白质发生交叉反应。有些错义突变不影响蛋白质或酶的生物活性,因而不表现出明显的表型效应,这种突变可称为中性突变(neutral mutation)。
3.无义突变当单个碱基置换导致出现终止密码子(UAG、UAA、UGA)时,多肽链将提前终止合成,所产生的蛋白质(或酶)大都失去活性或丧失正常功能,此种突变称为无义突变(non-sense mutation)。例如,DNA分子模板链中ATG的G被T代替时,相应的mRNA上的密码子便从UAC变成终止信号UAA,因此翻译便到此为止,使肽链缩短。无义突变如果发生在靠近3’末端处,它所产生的多肽链常有一定的活性,表现为渗漏型,这类多肽多半具有野生型多肽链的抗原特异性。
4.终止密码突变当DNA分子中一个终止密码发生突变,成为编码氨基酸的密码子时,多肽链的合成将继续进行下去,肽链延长直到遇到下一个终止密码子时方停止,因而形成了延长的异常肽链,这种突变称为终止密码突变(termination codon mutation),这也是种延长突变(elongtionmutation)。
5.抑制基因突变当基因内部不同位置上的不同碱基发生了两次突变,其中一次抑制了另一次突变的遗传效应,这种突变称为抑制基因突变(suppressor gene mutation)。例如Hb Harlem是β链第6位谷氨酸变成缬氨酸,第73位天冬氨酸变成天冬酰胺;如果单纯β6谷氨酸→缬氨酸,则可产生HbS病,往往造成死亡。但Hb Harlem临床表现却较轻,即β73的突变抑制了β6突变的有害效应。
(二)移码突变
移码突变(frame-shiftmutation)是指DNA链上插入或丢失1个、2个甚至多个碱基(但不是三联体密码子及其倍数),在读码时,由于原来的密码子移位,导致在插入或丢失碱基部位以后的编码都发生了相应改变。移码突变造成的肽链延长或缩短,取决于移码终止密码子推后或提前出现。(图3-11)
(三)整码突变
如果在DNA链的密码子之间插入或丢失一个或几个密码子,则合成的肽链将增加或减少一个或几个氨基酸,但插入或丢失部位的前后氨基酸顺序不变,称为整码突变(codon mutation)或密码子插入或丢失(codon insertion ordeletion)(图3-11)。
(四)染色体错误配对不等交换
染色体错误配对不等交换(mispairedsynapsis and unequal crossing-over)减数分裂期间,同源染色体间的同源部分发生联会和交换,如果联会时配对不精确,会发生不等交换,造成一部分基因缺失和部分基因重复。这种突变常用解释大段多核苷酸的丢失和重复(图3-21)。
图3-12 错误配对和不等交换
三、调控基因突变对结构基因表达的影响
所有细胞都是全能核(携带全部遗传信息,但不是全部基因都有活性,所以必定有一种抑制某些基因活笥和启动另一些基因活性的机制。对于基因调控机制1961年Jacob与Monod对大肠杆菌的研究提出了乳糖操纵子假说(Lac operon hypothesis),认为基因的作用单位是操纵了(operon),它由一个操纵基因和相邻的结构基因构成,它们按一定的线性顺序排列,并产生一系列相关的酶。操纵基因可以启动全组结构基因的活性,但它又被调节基因激活或抑制。调节基因能合成一种物质(阻遏物),能抑制操纵基因,当调节基因起作用时,有关的结构基因不合成蛋白质,只有在阻遏物被一种特殊代谢物(诱导物)灭活后,调节基因在关闭的情况下,结构基因才起作用。真核细胞的基因调控还未完全阐明。如果这一模式能应用在人类,即假设有不止缺乏一种相关酶的那些遗传病,有可能是由于调控系统基因突变的结果。又如有些酶活性缺乏或增加,或蛋白质合成量有改变,但从结构基因水平并未发现有任何碱基改变,这样推测突变可能发生调控基因部分,例如腺苷脱氨酶(adenosine deaminase,ADA)遗传性酶活性过高(相当于正常45-70倍)可引起溶血,但该突变酶结构没有改变,而转录的mRNA大大增多,故认为是调控基因突变的结果。又如Crigler-Najjar综合征Ⅱ型,表现为先天性黄疸,为肝葡萄糖醛酰转移酶缺乏,血中非结合胆红素增高。如用苯巴比妥可诱导此酶活性升高,黄疸消失,故认为此病可能是调节失控所致。
如果调节基因突变失去活性,基因不再被控制,结果蛋白质合成就会增加。在杂合子中,由于同源染色体上的正常调节基因所产生的阻遏物足够抑制两条染色体上的蛋白质合成,所以只有在纯合子才表现出来。另一种情况是,由于调节基因发生突变,合成了异常的阻遏物,它不能被诱导物所灭活,导致蛋白质合成减少。由于阻遏物是可以扩散的,它将有可能在同源染色体上起作用,因此杂合子即可表现出来。因此,尽管一般认为,遗传性代谢缺陷是由于结构基因突变造成的,但应考虑到调控基因突变也可引起表型相同的遗传病。近年来分子遗传学的发展,已从DNA顺序的改变证明了这一点,例如地中海贫血有些突变就发生在调控基因部分。
四、基因突变的后果
根据基因突变对机体影响的程度,可分为下列几种情况:
1.变异后果轻微,对机体不产生可察觉的效应。从进化观点看,这种突变称为中性突变。
2.造成正常人体生物化学组成的遗传学差异,这样差异一般对人体并无影响。例如血清蛋白类型、ABO血型、HLA类型以及各种同工酶型。但在某种情况下也会发生严重后果。例如不同血型间输血,不同HLA型间的同种移植产生排斥反应等。
3.可能给个体的生育能力和生存带来一定的好处。例如,HbS突变基因杂合子比正常的HbA纯合子更能抗恶性疟疾,有利于个体生存。
4.产生遗传易感性(genetic susceptibility).
5.引起遗传性疾病,导致个体生育能力降低和寿命缩短,这包括基因突变致蛋白质异常的分子病及遗传酶病。据估计,人类有50000个结构基因,正常人的基因座位处于杂合状态的可占18%,一个健康人至少带有5-6个处于杂合状态的有害突变,这些突变如在纯合状态时就会产生有害后果。
6.致死突变,造成死胎、自然流产或出生后夭折等。
第四章 单基因病
第一节 单基因病的遗传方式
单基因遗传病简称单基因病(monogenicdisease;single gene disorder)是指单一基因突变引起的疾病,符合孟德尔遗传方式,所以称为孟德尔式遗传病。
由于人类病症和性状不能如动物或植物那样通过杂交试验研究其遗传规律,因而必须采取适合于人类特点的研究方法。家系调查和系谱分析是判断某种遗传病遗传方式最常用的方法。系谱分析(pedigree analysis)是指将调查某患者家族成员所得到的该病或性状发生情况的资料,按一定格式绘制成图解(系谱)。对某病或性状遗传方式的判断必须进行多个系谱综合分析后方能作出准确结论。
绘制系谱图时采用统一的符号以表示家系中各个成员情况和相互之间的关系(图4-1)。
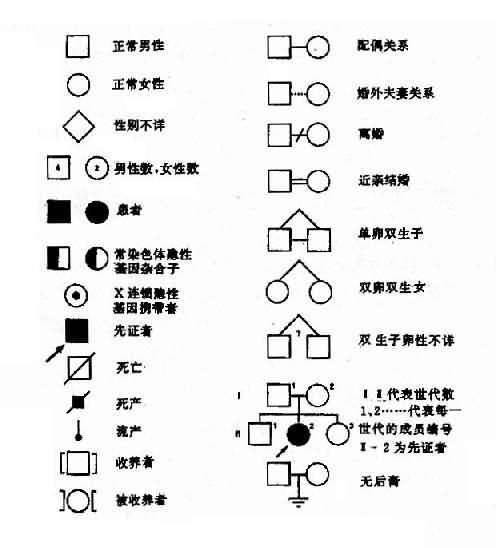
图4-1系谱符号
单基因病遗传方式的基本类型分述如下。
一、常染色体遗传
单基因涉及单个即一对等位基因发生突变所致的疾病,可按遗传方式分为下列4种主要类型。
1.由这种致病基因导致的疾病称为染色体显性遗传病。据统计,此类遗传病或异常性状已达3711种(1992年)。
致病基因有显性和隐性之分,其区别在于杂合状态(Aa)时,是否表现出相应的性或遗传病。若杂合子(Aa)能表现出与显性基因A有关的性状或遗传病时,其遗传方式称为显性遗传。
(1)完全显性遗传:凡是致病基因杂合状态(Aa)时,表现出像纯合子一样的显性性为状或遗传病者,称为完全显性(completedominance)。短指症(brachydactyly)可作为完全显性遗传的实例。本症为较常见的手(足)部畸畸形。由于指骨或掌骨变短,或指骨缺如,致使手指(趾)变短(图4-2)。
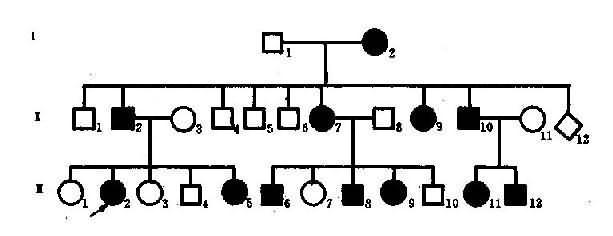
图4-2 一例短指(趾)症系谱
从系谱分析(图4-2),男女都可发病,与性别无关,所以本病是由某对常染色体上的基因决定的。假定A为显性基因(dominant gene),杂合状态时(Aa),只有基因A控制的性状显示出来,呈现出某种临床症状,而基因a的作用没有表达出来,称为隐性基因(recssive gene)。临床症状是表现出来的性状,称为表现型或表型(phenotype)。例如患者有短指,正常人没有短指,这是不同的表现型。控制各种表现型的遗传组成称为基因型或遗传型(genotype)。设短指症病人的基因型是Aa,正常人的基因型是aa.。等位基因Aa的人,两个基因不同,称为杂合子(heterozygote)。而一对基因相同(aa或AA)者,称为纯合子(homozygote)。因为A对a是显性的,基因A的作用在杂合子时表现出来,所以短指症的遗传方式是常染色体显性遗传。短指症的基因型有两种,纯合子(AA)和杂合子(Aa),它们在临床表现上无区别,故为完全显性。但临床上常见的情况都是杂合子患者(Aa)和正常人(aa)之间的婚配,后代中短指症患者与正常人的比例应为1:1,也就是说,后代将有约1/2子女发病,当两个短指症杂合子患者婚配时,其后代约3/4的子女将发病,只有约1/4子女正常。
图4-2的每个患者基因型都是杂合子(Aa),他(她)们的致病基因A一定来自双亲中的一方,所以双亲中的一方也是Aa,当然也是患者,这样就出现三代连续传递的现象。正常人的基因型都是aa,因此,患者的正常亲属也应都是aa,其子女都可能完全正常。该家系共21人,短指症患者11人(男5女6),发病比例接近1/2。应该指出,这种比例是在大样本的观察中方能反映出来,在子女数较少的小家庭往往不能反映出这种特点而出现较大的偏差。上述系谱基本反映了完全显性遗传特点,表现在:①连续四代发病;②患者子女中约1/2发病;③男女发病机会大致均等。
(2)不完全显性:有时杂合子(Aa)的表现型较纯合子轻,这种遗传方式称为不完全显性(incompletedominance)或半显性(semi-dominance),也称中间型遗传(intermedeate inheritance)。这里,杂合子(Aa)中的显性基因A和隐性基因a的作用都得到-定程度的表达。β地中海贫血可作不完全显性遗传实例,致病基因βO纯合子,基因型为βOβO者病情严重,杂合子基因型为βOβA者病情较轻,而正常基因βA纯合子基因型为βAβA者无症状。从临床症状轻重来看,杂合子βOβA病情是界于βOβO与βAβA之间(详见本章第三节)。
(3)共显性:一对常染色体上的等位基因,彼此间没有显性和隐性的区别,在杂合状态时,两种基因都能表达,分别独立地产生基因产物,这种遗传方式称为共显性遗传(co-dominace)。ABO血型的遗传可作为显性遗传的实例。ABO血型决定于一组复等位基因(multiple alleles)。复等位基因是指在一个群体中,一对特定的基因座位上的基因不是两种(如A和a),而是三种或三种以上,有时可达数十种。但是,对每一个人来说只能具有其中的任何两个等位基因。复等位基因是由于一个基因发生多种突变,从而产生多种基因型的结果。ABO血型的基因已定位于9q34,在这一座位上,由IA、IB、和i三种基因组成复等位基因。基因IA对基因i为显性,基因IB对基因i也是显性。基因型IAIA和IAi都决定红细胞膜上抗原A的产生,这种个体为A型血;基因型IBIB和IBi都决定红细胞膜上抗原B的产生,这种个体为B型血;基因型ii测定H物质的产生而不产生抗原A和抗原B。就IA、IB、i这一组复等位来说,复等位基因的数目是3个,所以共有:n(n+1)/2=3(3+1)/2=6
种基因型。在共显性时,有4种表现型。如果纯合子(IAIA)A型血的人与纯合子(IBIB)B型血的人结婚只能出生杂合子(IAIB)AB型血的子女;如果两个杂合子(IAIB)AB型血的人结婚则会导致1(IAIA):2(IAIB):1(IBIB)的比率,这样,3:1的比值就被1:2:1的比值所代替,这是两个等位基因共显性的结果。
根据孟德尔分离律的原理,已知双亲血型,就可以估计出子女中可能出现的血型和不可能出现的血型(表4-1),这在法医学的亲子鉴定中有一定意义。
表4-1双亲和子女之间血型遗传的关系
| 双亲的血型 | 子女中可能出现的血型 | 子女中不可能出现的血型 |
| A×A | A,O | B,AB |
| A×0 | A,O | B,AB |
| A×B | A,B,AB,O | - |
| A×AB | A,B,AB | O |
| B×B | B,O | A,AB |
| B×O | B,O | A,AB |
| B×AB | A,A,AB | O |
| AB×O | A,B | AB,O |
| AB×AB | A,B,AB | O |
| O×O | O | A,B,AB |
此外,人类MN血型、人类组织相容性抗原(human lecocyte antigen,HLA)系统等都是共显性例子。
(4)不规则显性:带有显性基因的个体理应发病,但事实上并非完全如此,有些杂合子(Aa)并不发病,这可能是因受修饰基因等因素的影响而不表现出临床症状,失去显性特点而不外显,有时表现程度有差异,称为不规则显性(irregular dominance)。修饰基因(modifier gene)是指本身没有表型效应,可是能对主基因发生影响,使主基因的表型成完全或能削弱主基因的作用,从而出现各种表现度和不完全的外显率。
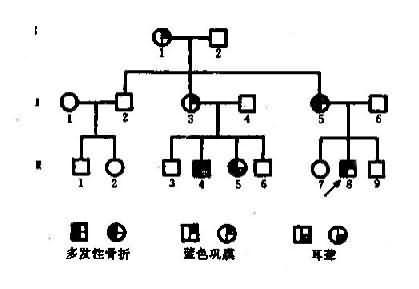
图4-3 一例成骨不全病例系谱
图4-3是一个成骨不全症的系谱,该家族中的患者有一共同的致病基因(A),均同I1传递而来,然而他们的临床表现却有很大的差别。先证者Ⅲ8有蓝色巩膜和多次骨折,其母亲Ⅱ5只有一次骨折史,其姨母Ⅱ3和外祖母Ⅰ1都只有蓝色巩膜,其姨表兄Ⅲ5则具多次骨折和耳聋两种症状,可见这些患者存在着明显的表现程度不一致。杂合子(Aa)在不同遗传背景和环境因素影响下,性状表现程度的差异称为表现度(expressivity)。上述种患者症状表现速度的区别可以解释为:Ⅰ1.Ⅱ3由于遗传背景中可能存在着减弱基因(reducer gene)作用,所以表现度轻:Ⅲ4 、Ⅲ5和Ⅲ8的遗传背景中可能存在增强基因(enhancergene),所以表现度重。
(5)延迟显性:有些显性遗传病并非出生后即表现出来,而是到较晚期才出现症状,这种情况称为延迟显性(delayed dominance)。慢性进行性舞蹈病(Huntington’schorea)可作为实例。此病为染色体显性遗传病,致病基因位于4p16。杂合子(Aa)在20岁时只有1%发病,40岁有38%发病,60岁有94%发病。这里,年龄对发病是一个重要的修饰因素。可见本病杂合子要个体发育早期,致病基因并不表达,但到一定年龄后,致病基因的作用方表达出来,主称为延迟显性。
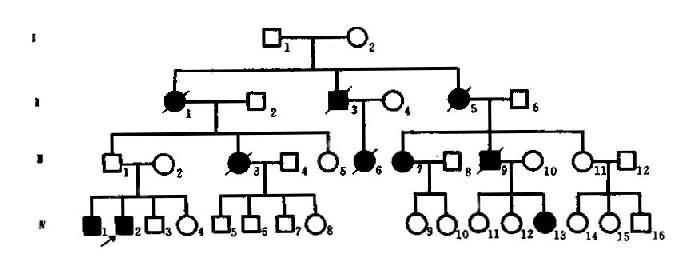
图4-4 例慢性进行舞蹈病系谱
图4-4是一例慢性进行性舞蹈病系谱。Ⅲ1未见发病,但他的母亲(Ⅱ1)和2个儿子(Ⅲ1,2)均已患病,因此可以认为Ⅲ1携带有致病基因,由于某种原因未能表现症状,因而出现了隔代传递现象。显性基因完全不能表达的个体称顿挫型(form fruste)。Ⅲ1是顿挫型,虽未发病的,便仍将致病基因传给后代。因此,本例是不规则显性。顿挫型的存在形成致病基因(A)不完全外显,这样显性基因在杂全状态时是否得到表现,可用外显就绪来衡量。外显率(penetrance)是指显性基因能形成相应表现型的比例,一般用百分率(%)来表示。显性基因能100%表现出相应性状称为完全外显,外显率低于100%时为不完全外显或外显不全。一般外显率高者可达70%-80%,低者只有20%-30%。当计算外显率时应搜集较多的家系汇总分析方能符合实际情况。
延迟显性的一特点是,最年轻一代的患者比例常不足1/2。图4-4中的第Ⅳ代患者仅3/13即为佐证。
2.常染色体隐性遗传控制遗传性状或遗传病的基因位于常染色体上,其性质是隐性的,在杂合状态时不表现相应性状,只有当隐性基因纯合子(aa)方得以表现,称为常为常染色体隐性遗传病(autosomal recessiveinheritance,AR)。这种致病基因所引起的疾病称为常染色体隐性遗传病。目前已知的常染色体隐性遗传病或异常性状达1631种(1992年)。白化病(albinism)可作为常染色体隐性遗传病的实例。白化病是由于全身黑色素细胞均缺乏黑色素,所以皮肤毛发呈白色。本病患者只有当一对等位基因是隐性致病基因纯合子(aa)时才发病,所以患者的基因型都是纯合子(aa)。当一个个体为杂合状态(Aa)时,虽然本人不发病,但为致病的基因携带者,他(她)能将致病基因a传给后代,因此患者父母双方都应是致病基因(Aa)的肯定携带者(obligatory carrier)。如果两个杂合子(Aa)婚配,后代子女患者(aa)占1/4,表型正常者占3/4。表型正常的人中1/3基因型为纯合子(AA),2/3为杂合子(Aa),是致病基因的可能携带者(probable carrier)。
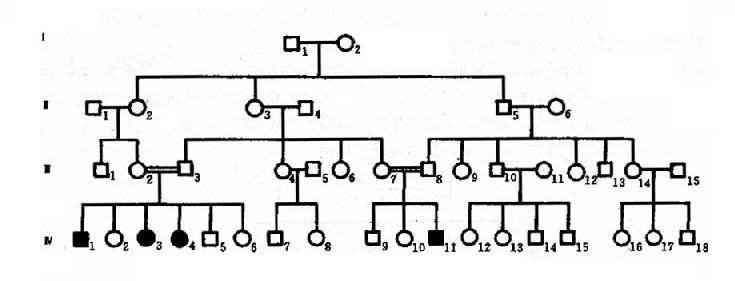
图4-5 一例白化病系谱
图4-5是白化病的一个家系,这个系谱基本反映了常染色体隐性遗传的特点。表现在:①患者(Ⅳ1,3,4,11)的双亲(Ⅲ2,3和Ⅲ7,8)表现型正常,但均为致病基因的肯定携带者;②系谱中看不到连续遗传现象,常为散发,有的系谱中只见先证者;③同胞中约1/4个体发病,男女性发病机会均等,患者大部分出现在同胞之间,子女往往正常;④近亲婚配的后代发病概率显著较高,系谱中的Ⅲ2和Ⅲ3.Ⅲ7和Ⅲ8都是近亲婚配。
二、性染色体遗传
性染色体上的基因所控制的遗传性状或遗传病,在遗传上总是和性别相关的。目前已知的性连锁遗传的致病基因大都在X染色体上,与性别相关联的遗传方式称为性连锁遗传(sexlinkedinheritance)。目前已知的X连锁隐性遗传病或异常性状有360种(1992年)。
1.X连锁隐性遗传一种性状或遗传病有关的基因位于X染色体上,这些基因的性质是隐性的,并随着X染色体的行为而传递,其遗传方式称为X连锁隐性遗传(X-linked recessive inheritance,XR).
以隐性方式遗传时,由于女性有两条X染色体,当隐性致病基因在杂合状态(XAXa)时,隐性基因控制的性状或遗传病不显示出来,这样的女性表型正常的致病基因携带者。只有当两条X染色体上等位基因都是隐性致病基因纯合子(XaXa)时才表现出来。在男性细胞中,只有一条X染色体,Y染色体上缺少同源节段,所以只要X染色体上有一个隐性致病基因(XaY)就发病。这样,男性的细胞中只有成对的等位基因中的一个基因,故称为半合子(hemizygote)。
红绿色盲可作X连锁隐性遗传病实例。色盲有全色盲(achromatopsis)和红色绿色盲(dyschromatopsia of theprotan and deutan)之分。前者不能辨别任何颜色,一般认为是常染色体隐性遗传;后者最为常见,表现为对红绿色的辨别力降低。呈X连锁隐性遗传,致病基因定位于Xq28。据报道,男性发生率7.0%,女性为0.5%.一个红绿色盲男患者(XbY)和正常辩色能力女性(XBXB)结婚,他们的女儿都应从父亲那里接受一个X染色体,从母亲那里得到一条正常的X染色体而成为致病基因携带者杂合了(XBXb),他们的儿子必定由母亲那里接受一条XB,故辩色能力全部正常(XBY)。凡携带致病基因的女性(XBXb)与正常辩色男人结婚,下一代中,儿子有一半是正常(XBY)的,一半是绿色盲(XbY),女儿中一半是致病基因携带者(XBXb),一半则完全正常(XBXB)。因此,男性患者的父亲一定是患者,其母亲是致病基因携带者。这里可见“父传女,母传子”的交叉遗传(criss-crossinheritance)现象,如果女性携带者(XBXb)与男性患者(XbY)s结婚,后代中,女儿1/2可能发病,1/2可能为携带乾,独生子中发病者和正常者各占1/2。
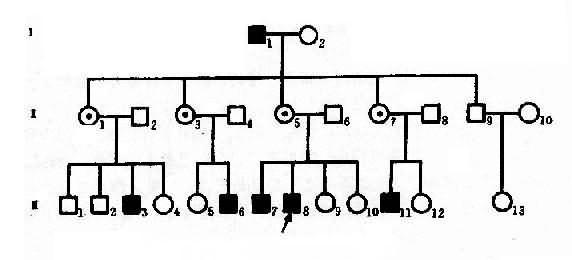
图4-6 一例红绿色盲系谱
从图4-6红绿色盲系谱分析,男性红绿色盲患者I1(XbY)和正常辩色能力女性I2结婚,他们的女儿全部是杂合子(Ⅱ1,3,5,7),因为儿了(Ⅱ9)只能从母亲处得到XB,故表型正常,在下一代中,携带致病基因的女性(XBXb)与正常男性(XBY)结婚时,他们的儿子有1/2可能是正常的(XBY),1/2可能是红绿色盲(XbY)(Ⅲ3,6,7,8,11),女儿中1/2可能是携带者(XBXb),1/2可能完全正常(XBXB),这样出现代与代间明显的间隔遗传现象。该系谱先证者Ⅲ8的妹妹Ⅲ9,10姨表姐妹Ⅲ4,5,12,-14虽表型正常,但有1/2可能是携带者,她们结婚后也有可能把致病基因传给儿子。
从红绿色盲系谱中,可反映出X连锁隐性遗传系谱和特点,表现在:①男性患者过远多于女性患者,系谱中的病人几乎都是男性;②男性患者的双亲都无病,其致病基因来自携带者母亲;③由于交叉遗传,男患者的同胞、舅父、姨表兄弟、外甥中常见到患者,偶见外祖父发病,在此情况下,男患者的舅父一般正常;④由于男患者的子女都是正常的,所以代与代间可见明显的不连续(隔代遗传)。
2.X连锁显性遗传一些性状或遗传病的基因位于X染色体上,其性质是显性的,这种遗传方式称为X连锁显性遗传(X-linked dominant inheritance),这种疾病称为X连锁显性遗传病。目前所知X连锁显性遗传病不足20种。
由于致病基因是显性的,并位于X染色体上,因此,不论男性(XAY)和女性(XAXa)只要有一个这种致病基因XA就会发病。与常染色体显性遗传不同之处是,女性患者既可将致病基因传给生子,又可以传给女儿,且机会均等;而男性患者只能将致病基因传给女儿,不传给儿子。由此可见,女性患者多于男性,大约为男性的1倍。另外,从临床上看,女性患者大多数是杂合子,病情一般较男性轻,而男患者病情较重。
抗维生素D佝偻病(vitamin D resistant rickets, VDRR)可以作为X连锁显性遗传病的实例。VDRR是一种以低磷酸血症导致骨发育障碍为特征的遗传性骨病。患者主要是肾远曲小管对磷的转运机制有某种障碍,困而尿排磷酸盐增多,血磷酸盐降低而影响骨质钙化。患者身体矮小,有时伴有佝偻病等各种表现。患者用常规剂量的维生素D治疗不能奏效,故有抗维生素D佝偻病之称。从临床观察,女性患者的病情较男性患者轻,多数只有低血磷,佝偻症状不太明显,表现为不完全显性,这可能是女性患者多为杂合子,其中正常X染色体的基因还发挥一定的作用。
男性患者(XHY)与正常女性(XhXh)结婚,所生子女中,儿子全部正常,女儿全部发病;女性患者(XHXh)与正常男性(XhX)结婚,子女中正常与患者各占1/2。
图4-7是抗维生素D佝偻病系谱,女性患者I1(XHXh)产生两种配子,她与正常男性结婚,理论上子女正常与患者各占1/2,故Ⅱ1、Ⅱ2、Ⅱ3都可能发病,Ⅱ3的子女Ⅲ12,13,14也可能发病;但男性患者与正常女性结婚,由于男性患者把致病基因只传给他的女儿,不传给儿子,所以Ⅲ12和Ⅲ14的女儿(Ⅳ18,Ⅳ21)都发病,儿子(Ⅳ19,Ⅳ20)正常。同时可见到上代传给下代的连续性。女性(Ⅲ3、4、6)患者只有低血磷,没有佝偻病。本系谱可以反映出X连锁显性遗传特点,表现在:①女性患者多于男性,女性患者(Ⅲ3、4、6)病情较轻,只有低血磷;②患者双亲之一必定是患者,女患者都是杂合子,她们的致病基因可传给儿子和女儿,但男患者的致病基因只传给女儿,因此系谱中男患者的女儿全部发病;③可看到连续两代以上都有患者。
3.Y连锁遗传如果致病基因位于Y染色体上,并随着Y染色体而传递,故只有男性才出现症状。这类致病基因只由父亲传给儿子,再由儿子传给孙子,女性是不会出现相应的遗传性状或遗传病,这种遗传方式称为Y连锁遗传(Y-linked inheritance)。由于这些基因控制的性状,只能在雄性个体中表现,这种现象又称为限雄遗传(holandric inheritance)。
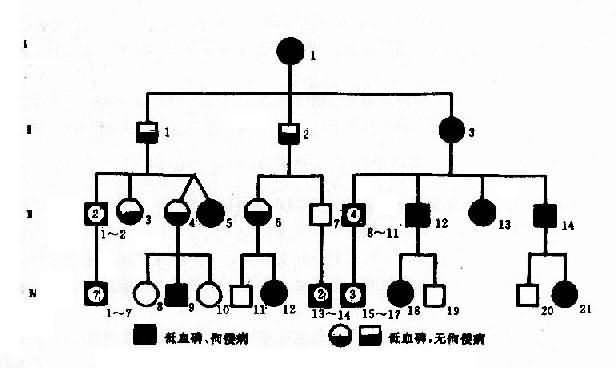
图4-7 一例抗维生素D佝偻病系谱
迄今报道Y连锁遗传病及异常性状仅10余种。我国发现一个视网膜色素变性的家系,4代共26人中,8例患者均为男性,女性正常且后代亦无患者,很可能属Y连锁遗传,有待进一步证实。另外耳毛性状呈Y连锁遗传较多见。
除上述几种基本遗传方式外,尚有2种特殊情况:
(1)从性遗传:从性遗传睡性连锁遗传的表现都与性别有密切关系,但它们是两种截然不同的遗传方式。性连锁遗传的基因位于性染色体上,而从性遗传的基因位于常染色体上,致病基因性质有显性和隐性之别。这种常染色体上的基因所控制的性状,在表现型上受性别影响而男女性分布比例或表现程度上的差别,这种遗传方式称为从性遗传(sex-influrenced inheritance)。
原发性血色病(primaryhematochromatosis)可作从性遗传方式的实例。本病为一种遗传性铁代谢障碍,其特征为含铁血黄素在组织中大量沉积,造成多种器官损害,典型症状是皮肤色素沉着、肝硬化、糖尿病三联综合征,症状发生较迟,由于铁质蓄积达到15-30g方产生症状,所以80%病例在40岁以后发病。本病致病基因在常染色体上,但男性多于女性10-20倍,而且女性发病较迟,这是因为女性通过月经、妊娠和哺乳,一生顺可丧失铁10-35g,故难以表现铁质沉着症状。
遗传性早秃(hereditaryalopecia)为常染色体显性遗传病,男性显著多于女性,女性仅表现为头发稀疏,极少全秃,杂合子(Bb)男性会出现早秃;相反,女性杂合子(Bb)不出现早秃,只有纯合子(BB)才出现早秃,这也是从性遗传的一例。
(2)限性遗传;一种遗传性状或遗传病的致病基因位于常染色体或性染色体上,其性质可以是显性或隐性,但由于性别限制,只在一种性别得以表现,而在另一性别完全不能表现,但这些基因都可以向后代传递,这种遗传方式称为限性遗传(sex-limited inheritance)。例如,子宫阴道积水(hydrometrocolpos)由常染色体隐性基因决定,因此,女性只有在纯合子才表现相应症状,男性虽有这种基因但不能表现该性状,然而这些基因都向后代传递。
上述从性遗传和限性遗传特点可见,并非所有表现出性别差异的遗传性状或遗传病都是性连锁遗传,在常染色体遗传病中有时也可见到性别差异,应注意加以区别。
三、两种单基因病或性状的遗传规律
1.两种单基因病的致病基因分别位于不同对染色体上,在临床上,一个家系中如果出现两种单基因病患者,在大多数情况下,其遗传方式受孟德尔自由组合律制约。假设一个并指症(并指完全,伴有掌、跖骨融合)女性与一个甲型血友男患者结婚,生育了一个男孩,其并指症状与母亲类同,他们要求再生一孩子,试问后代子女发病风险如何?
并指症是常染色体性遗传病,因此母亲的基因型为XXBb,甲型血友病是X连锁隐性遗传病,男性是半合子发病,所以父亲的基因型是XhYbb。从图4-8可以看出,他们后代的女孩中,有50%可能患并指症伴甲型血友病携带者,50%可能是甲型血友病携带者;男孩中,50%可能是并指症,但有50%可能正常。
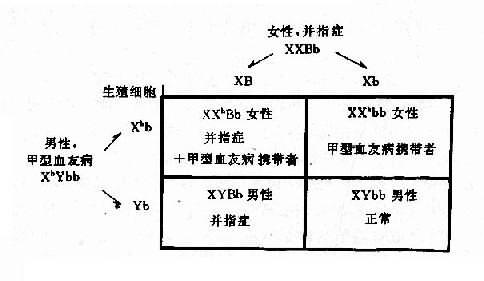
图4-8 两种致病基因在不同染色体上的自由组合
B:并指症基因;h:甲型血友病基因
2.两种单基因病基因位于同一染色体上有时患者有两种单基因遗传病,如果这两种致病基因位于同一染色体上,它们将表现为连锁遗传,其遗传方式受连锁与交换律制约。例如,红绿色盲与甲型血友病的基因都是在X染色体上,所以彼此连锁。假定两者间交换率是10%。如果父亲是红绿色盲,母亲外表正常,已生出一个女儿是红绿色盲,一个儿子是甲型血友病,试问他们以后所生的孩子中,这两种遗传病的发病风险如何?能生出正常的后代吗?
从一个女儿是红绿色盲来看,母亲必然是红绿色盲基因(b)的携带者,再从一个儿子是甲型血友病来看,母亲也必然是甲型血友病基因(h)的携带者。但是,这两种致病基因分别位于两条染色上。父亲是红绿色盲患者,所以有红绿色盲基因(b)。从图4-9可以看出,他们后代的女孩中,50%可能是正常的,50%可能患红绿色盲;男孩中,45%可能患红绿色盲,45%可能患甲型血友病,5%可能同时患这两种病,只有5%可能是正常的。
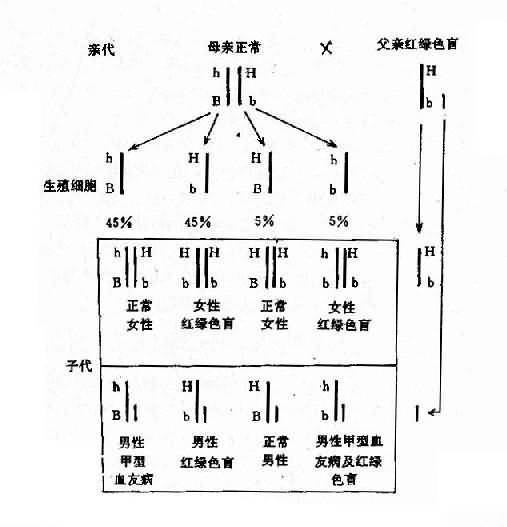
图4-9 两种X连锁隐性遗传病的连锁和交换
h:甲型血友病基因:b:红绿色盲基因
综上所述,研究两种基因病伴随遗传规律,在遗传咨询时估测遗传病患者后代发病风险是重要的。
3.两对基因的相互作用先天性聋哑是一个较常见的两对基因相互作用的例子。假定在不同座位上的两套以上的隐性基因中,只要任何一个座位是隐性纯合子,就出现聋哑。
(1)聋哑双亲生育的子女可以是全部聋哑或全部正常
DDee×DDee→DDee
ddEE×ddEE→ddEE
DDee×ddEE→DdEe 全部正常
(2)不同家系的聋哑尊重亲生育正常听觉子代相互结婚,有较高机会出现聋哑儿女
即正常听觉占9/16,聋哑占7/16
四、单基因病的遗传异质性与遗传方式
遗传异质性(heterogeneity)是指表现型一致的个体或同种疾病临床表现相同,但可能具不同的基因型,称为遗传异质性。由于遗传基础不同,它们的遗传方式、发病年龄、病程进展、病情严重程度、预后以及复发风险等都可能不同。研究表明,遗传病病种增多的原因不仅是由于发现了新的疾病,而是从已知的综合征中分出了亚型,即遗传异质性的存在。遗传异质性几乎成为遗传的普通现象。例如视网膜色素变性(retinitis pigmentosa,RP)是最常见的致盲的单基因遗传眼病之一,主要表现为视网膜萎缩、夜盲和视野缩小,多为双眼发病,致中年或老年进完全失明。
RP的遗传方式具有遗传异质性,即可以有AD、AR、XR连锁遗传,可能还有Y连锁遗传。遗传方式不同的RP,一般其遗传基础也不同,因而伴随的综合征的以及始发年龄、主要病情变化特征(XR常伴高度近视,AR和AD多为低度近视)、病程进展(AD快,AR慢)、预后情况(AD较轻,AR致盲)也有差异,甚至还可区分为其他不同亚型。
现知,XL的RP2基因定位于Xp11.4-11.23,XL的PR3基因定位于Xp21.1,AD的RP基因定位于8p11-q21。因此,普遍认为RP是多个基因座位上RP基因所引起的一组具有临床亚型的视网膜退行性病变的遗传性疾病。
五、不同于孟德尔遗传规律的遗传现象
1.母系遗传母系遗传(maternalinheritance)是指核外染色体所控制的遗传现象。例如Leber遗传性视神经病(Leber’s heredi tary optic neuropathy,LHON),也称Leber病。其主要病变为视神经退行性变,发病较早,表现为急性亚急性视力减退,中心视野丧失最明显。此病发病机制一般认为是由于mtDNA点突变导致其第11778位精氨酸→组氨酸(多见)及细胞色素b第15257位天冬氨酸→天冬酰胺。前者使编码呼吸链NADH脱氢酶mtDNA第340位精氨酸被组氨酸取代,改变了mtDNA阀间构型,导致NADH脱氢酶活性降低,线粒体产能下降,因而对需能量多的视神经组织损害最大,久之导致视神经细胞退行性变,直至萎缩。
由于mtDNA为母系遗传,因此由mtDNA基因突变所致的Leber病也遵循母系遗传的传递规律,即患者都与母亲有关。男性患者的后代中尚未见有直接传代者。但并非女性患者的后代全部发病,而且发病年龄也不一致;甚至一些女性患者本身表型正常,但可将本病传给下一代。母系遗传的特点:①母亲将她的mtDNA传递给儿子和女儿,但只有女儿能将其mtDNA传递给下一代;②人的细胞里通常有上千个mtDNA拷贝,在突变体和正常mtDNA共存的细胞中,mtDNA在细胞的复制和分离过程中发生遗传漂变,可导致子细胞出现三种基因型:纯合的突变体mtDNA、纯合的正常mtDNA、突变体和正常的mtDNA的杂合,这是由于mtDNA的遗传不遵循孟德尔定律,被随机分配到子细胞中所致;线粒体病发病有一阈值,只有当异常的mtDNA超过阈值时才发病。女性携带者的细胞内突变的mtDNA未达到阈值或在某种程度上受核影响而未发病,但仍可以通过mtDNA突变体向下代传递。女性患者细胞里mtDNA同样可能存在杂合性,子女中得到较多突变mtDNA的个体发病,得到较少的病情较轻或不发病。
2.遗传印记根据孟德尔的遗传定律,当一个性状从亲本传给子代,无论携带这个性状的基因或染色体来自父方或母方,所产生的表型效应是相同的。但是目前发现同一种染色体(或基因)的改变由于不同性别的亲本传给子女时可以引起不同的疾病。例如,Prader-Willi综合征(PWS)和Angelman综合征(AS)是两种不同的遗传病,但都有共同的15q11-13缺失。父源染色体缺失时临床上为PWS,而母源染色体缺失时表现为AS。这提示来源不同的等位基因有不同的表达。某些常染色体显性遗传病的发病年龄和病情轻重似乎与传递基因亲本有关。慢性进行性舞蹈病患者发病年龄一般在30-50岁,但有5%-10%患者在20岁以前发病,且病情严重,这些患者致病基因均由父亲遗传。母亲遗传者,子女发病年龄多在40-50岁。囊性纤维化(cystic fibrosis,CF)是一种常染色体隐性遗传病,已发现某些CF患者的二条7号染色体均来自母亲,即单亲二体性(uniparental disomy,UPD)。人类的胚胎发育也有类似现象,拥有父源两套染色体的受精卵发育成葡萄胎,而拥有母源两套染色体的发育成卵巢畸胎瘤。此外,无论是双雄三倍体还是双雌三倍体都发育成畸胎儿。因此,正常的胚胎发育必须拥有亲代双方染色体或基因组。一些胚胎性肿瘤中也存在亲源性非随机的染色体或基因丢失现象,而且主要是母源染色体的丢失。如散发的肾母细胞瘤(Wilms trmor)有11p13-15的基因丢失,且皆来自母方,而遗传型基因丢失多来自父方;遗传型视网膜母细胞瘤(Rb)中有13q14杂合性丢失(LOH),且丢失的多为母系源Rb基因(详第九章)。
目前已知,至少有数十种遗传病存在着遗传印记现象,这种现象很难用经典的孟德尔定律来解释,也不能用性连锁遗传、线粒体遗传及多基因遗传来回答。近年来,揭示了一种新的遗传现象,即基因组印记(genomic imprinting),亦称遗传印记(geneticimprinting),是指来自双亲的基因或染色体存在着功能上的差异,因而子女来自父方与来自母方的基因表达可以不同。这是由于基因在生殖细胞分化过程中受到不同修饰的结果。换言之,遗传印记是一种依赖于配子起源的某些等位基因的修饰现象.一些基因在精子生成过程中被印记,另一些基因在卵子生成过程中被印记,被印记了的基因,它们的表达受到抑制。
遗传印记现象已在哺乳动物和人类中确认,但对印记现象的机理仍了解很少。据推测DNA的甲基化可能遗传印记的分子机理之一。在精子和卵子中一些基因甲基化程度不同,高度甲基化(被印记)的基因不表达或表达程度降低,当胚胎发育过程中发生去甲基化时,这些基因即开始表达。总之,基因的印记影响到性状或许多遗传病和肿瘤的发生,影响发病年龄、外显率、表现度,甚至遗传方式。在对某些不能用经典孟德尔定律解释的遗传现象时,用遗传印记可以得到合理解释。
第一节 基因突变致蛋白质合成异常
蛋白质性质是由DNA分子上碱基数量和顺序决定的。如果DNA分子的碱基数量或顺序发生变化,由它编码的蛋白质结构就发生相应的改变。由于基因突变导致蛋白质分子质和量异常,从而引起机体功能障碍的一类疾病称为分子病(molecular disease)。
分子病种类很多,根据各种蛋白质的功能可将分子病分为运输性蛋白病、凝血及抗凝血因子缺乏症、免疫蛋白缺陷病、膜蛋白病、受体蛋白病等.
一、血红蛋白病
血红蛋白病(hemoglobinopathy)是指由于珠蛋白分子结构或合成量异常所引起的疾病。它是人类孟德尔或遗传病中研究得最深入、最透彻的分子病,是运输性蛋白病的代表,是研究人类遗传机理的最好模型。据估计,全世界有一亿多人携带血红蛋白病的基因,我国南方发病率较高,因此,血红蛋白病是最常见的遗传之一。
(一)正常血红蛋白的组成,结构及遗传控制
1.人类血红蛋白的组成和发育变化 每个红细胞内含有约28000万个血红蛋白分子,每个分子由四个亚单位构成,每一个单位由一条珠蛋白肽链和一个血红素辅基组成,即血红蛋白分子是由二对珠蛋白链构成的球形四聚体(图4-10)。其中一对是类α链(α链和ξ链),由141个氨基酸组成;另一对是类β链(ε、β、γ和δ链),由146个氨基酸组成。由这6种不同的珠蛋白链组合成人类的6种不同的血红蛋白,即Hb Gower1(ξ2ε2)、HbGower2、(α2ε2)、HbPortland(ξ2γ2)、HbF(α2γ2)、HbA(α2β2)和HbA2(α2δ2)。其中γ链有两种亚型,即Gγ2和Aγ2,因此HbF有两类:α2Gγ2和α2Aγ2,前者的第136位氨酸为甘氨酸,后者为丙氨酸。
上述各种血蛋白在发育的不同阶段先后交替出现(图4-11)。在胚胎发育早期,合成胚胎血红蛋白HbGowerl、HbGower2和HbPortland。胎儿期(从8周至出生为止)主要是HbF。成人有3种血红蛋白:HbA,占95%以上;HbA2,占2%-3.5%;HbF,少于1.5%。
2.人类珠蛋白基因人类珠蛋白基因分为两类:一类是类α珠蛋白基因簇(α-likeglobin gene cluster),包括ξ和α基因;另一类是β珠蛋白基因簇(β-like globin gene cluster),包括ε、γ(Gγ和Aγ)、δ和β基因。
(1)类α珠蛋白基因:人类α珠蛋白基因簇位于16p13,每条染色体上均有两个α珠蛋白基因,因此,二倍体细胞中共有4个α基因,每个α基因几乎产生等量的α珠蛋白链。此外,在类α珠蛋白基因簇中,还包括两个ξ基因和一个假基因Ψα,这些基因紧密连锁其排列顺序如图4-12所示。
图4-10 血红蛋白结构示意图
A:血红蛋白四聚体(四级结构)
B:血红蛋白单体的三维空间结构
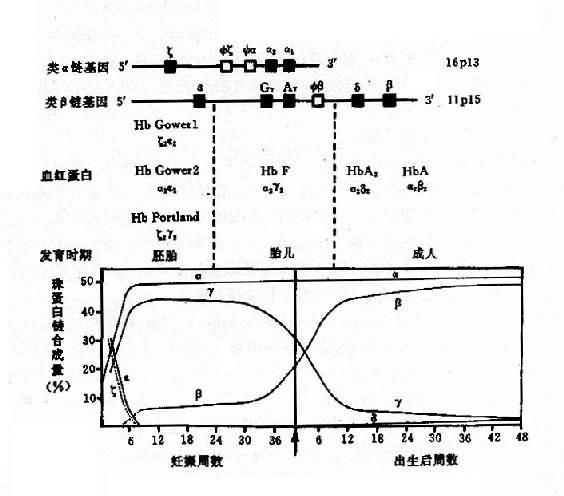
图4-11 人类血红蛋白类型及基发育过程中的演变
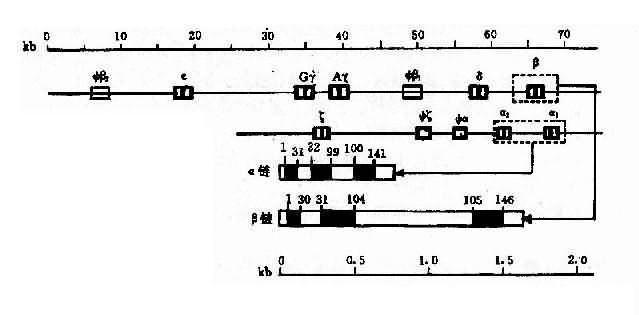
图4-12 人类α珠蛋白基因簇和人类β珠蛋白基因簇的结构及排列顺序
(2)类β珠蛋白基因:人类β珠蛋白基因簇分布于11p15,每条11号染色体上只有一个β珠蛋白基因,基因簇内各成员也都紧密连锁,其排列顺序如图4-12。类β珠蛋白基因簇的排列顺序与以育过程中的表达次序完全一致。
(3)珠蛋白基因的结构:类α与类β珠蛋白基因的结构相似,都含有3个外显子和2个内含子(IVS2和IVS2)。α珠蛋白基因中的IVS1和117bp组成,位于31和32密码子之间,IVS2由149或140bp组成,位于99和100密码子之间。类β基因中的IVS1含130bp,位于30和31密码之间,IVS2大约含850bp,位于104和105密码子之间。
(二)血红蛋白病的分类和分子基础
血红蛋白病可分为两大类,即异常血红蛋白病和地中海贫血。
1.异常血红蛋白病异常血红蛋白(abnormalhemoglobin)是指由于珠蛋白基因突变导致珠蛋白肽链结构异常,如有临床表现者称为异常血红蛋白病或异常血红蛋白综合征。至今全世界已发现异常血红蛋白471种。国内已发现60种,其中20种是世界首报。尽管异常血红蛋白种类繁多,但仅约40%的异常血红蛋白对人体有不同程度的功能障碍。
(1)异常血红蛋白病的类型:
1)镰形细胞病(sickle cell disease):此病主要见于黑人。该病系由于β链第6位谷氨酸被缬氨酸取代,形成HbS,导致电荷改变,在脱氧情况下HbS聚合形成长棒状聚合物,使红细胞镰变,由于镰变引起血粘度增高,导致血管梗阻性继发症状,一过性剧痛(肌肉骨骼痛、腹痛),急性大面积组织损伤,心肌梗塞可致死,镰变细胞的变性降低还可引起溶血。HbS纯合子(HbSHbS)表现为镰形细胞性贫血,杂合子(HbAHbS)表现为镰形细胞性状,大部分无症状,但也可有轻度慢性贫血。
2)不稳定血红蛋白病(unstable hemoglobinpathies):已发现的不稳定血红蛋白在80种以上。由于Hb不稳定容易自发(或在氧化剂作用下)变性,形成变性珠蛋白小体(Heinz小体)。Heinz小体粘附红细胞膜上,导致了离子通透性增加;另外,由于变形性降低,当红细胞通过微循环时,红细胞被阻留破坏,导致血管内、外溶血。不稳定Hb病一般呈常染色体显性遗传(不完全显性),杂合子可有临床症状,纯合子可致死。临床表现与Hb不稳定程度、产生高铁血红蛋白的多少以及不稳定Hb的氧亲和力大小有关。轻者仅在服用磺胺等药物或有感染时溶血;重者需反复输血才能维持生命。
3)血红蛋白M病(HbM):HbM是因肽链中与血红素铁原子连接的组氨酸或邻近的氨基酸发生了替代,导致部分铁原子呈稳定的高铁状态,从而影响了正常的带氧功能,使组织供氧不足,导致临床上出现紫绀和继发性红细胞增多。本病呈常染色体显性遗传,杂合子HbM含量一般在30%以内,可引起紫绀症状。
4)氧亲和力改变的血红蛋白病:这类血红蛋白病是指由于肽链上氨基酸替代而使血红蛋白分子与氧的亲和力增高或降低,致运输氧功能改变。如引起Hb与氧亲和力增高,输送给组织的氧量减少,导致红细胞增多症;如引起Hb与氧亲和力降低,则使动脉血的氧饱和度下降,严重者可引起紫绀症状。
(2)异常血红蛋白的分子基础:异常血红蛋白的发生涉及基因突变的各种类型,概括举例如下。
1)单个碱基置换:大多数异常血红蛋白是由于珠蛋白基因发生单个碱基置换所致,其中多为错义突变。
①错义突变:例如镰形细胞贫血是β基因第6位密码子GAG变成GTG。中国人较常见的HbE是β基因第26位密码子由GAG(谷)→AAG(赖)所致。
②无义突变:例如HbMckees-Rock,其β链只有144个氨基酸组成,原因是β基因第145位酪氨酸密码子TAT改变为终止密码子TAA,使肽链合成提前终止。
③终止密码突变:例如Hb Constant Spring就是由于α珠蛋白基因第142位终止密码子TAA(mtRNA为UAA)突变为CAA(谷氨酰胺),结果α延长为172个氨基酸,这种突变基因转形成的mRNAI不稳定,所以导致α链合成减少,表现为α+地中海贫血。
2)移码突变:例如Hb Wagne是由于α链第138位丝氨酸密码子UCC丢失一个C,致使其3’端碱基顺序依次位移,重新编码,第142位终止信号变为可读密码,致使翻译至147位才终止(图4-13)。
3)整码突变:例如Hb Gum Hiu是β链缺失第91-95氨基酸(亮-组-半胱-门冬-赖),但其前后氨基酸顺序正常(图4-13)。

图4-13 人类α链和β链mRNAR不同突变类型
4)融合基因:例如Hb Lepore的类β链是由δ链和β链连接而成,肽链的N端像δ链,C端像β链,故称为δβ链,相反,Hb反Lepore(Hbanti-Lepore)其N端像β链,C端却像δ链,称为βδ链。这是由于染色体的错配联会和不等交换而形成的融合基因(fusion gene)(图4-14)。
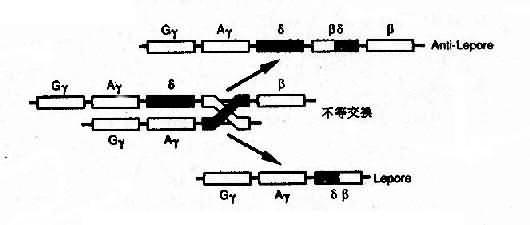
图4-14 血红蛋白融合基因形成机理
2.地中海贫血由于珠蛋白基因缺失或突变导致某种珠蛋的链合成障碍,造成α链和β链合成失去平衡面导致的溶血性贫血称为地中海贫血(thalassemia)。根据合成障碍的肽链不同可把地中海贫血分为α和β地中海贫血两类。此外还有少见的δβ和γβ地中海贫血。
(1)α地中海贫血(α-thalassemia,简称α地贫)是由于α珠蛋白基因的缺失或缺陷使α珠蛋白链(简称α链)的合成受到抑制而引起的溶血性贫血。如果一条16号染色体缺失1个α基因者称α+地贫(亦称α地2),缺失2个α基因者称为α0地贫(亦称α地1)。α地1的基因型可写作―/αα,α地2的单倍型则写成α-/αα.以上每种α地贫基因型与正常型配合可构成各种α地贫杂合子.各种α地贫基因型杂合子相互配合可构成各种纯合子或双重杂合子.α地中海贫血在我国多见于南方各省.
1)α地贫的临床类型:根据临床表现程度,依受累α基因数量不同而有差异,基本上可分为4类型.
①HbBart’s胎儿水肿综合征(HbBart’s hydrops fetalis syndrome):是两条16染色体的4个α基因全部缺失或缺陷,基因型为α0地贫纯合子(--/--),完全不能合成α链,不能形成胎儿HbF,相对过多的γ链形成γ四聚体(γ4)称为HbBart’s(γ4).HbBart’s对氧亲和力非常高,因而释放给组织的氧减少,造成组织严重缺氧导致胎儿水肿,引起死胎或新生儿死亡.患儿血红蛋白60%以上为HbBart’s,其余为 HbPortland.
患儿父母均为α0地贫杂合子,基因型为α/--.他们若再生育,则胎儿有1/4的机会为αHbBart’s水肿胎儿,1/4为正常人,1/2为α0地贫杂合子(α地1)
②血红蛋白H病:是α0地贫和α+地贫的双重杂合子,即有3个α基因缺失或缺陷,基因型为-α/--或α-/--,也可为ααT/--(αT代表有突变,如Hb Constant Spring)。因缺失3个α基因,只能合成少量α链,β链相对过多,形成β四聚体(β4),易被氧化,导致β4解体成游离的单链,游离β链沉淀聚积包涵体,附着于红细胞膜上,使红细胞膜受损,失去柔韧性,易被脾破坏,导致中等度或较严重的溶血性贫血,称为血红蛋白H病(Hbh disease) .
患者双亲的基因型多为α0地贫杂合子(αα/--)和α+地贫杂合子(α/αα),或为α0地贫杂合子(αα/--)和非缺失型地贫杂合子(ααT/αα)。婚配后其子女有1/4机会为正常人1/4为α+地贫杂合子,1/4为α0地贫杂合子及1/4为HbH病。如父母一方有αT,则导致非缺失型HbH病。
③轻型(标准型 )α地中海贫血:为α0地贫杂分子(--/αα)或α+地贫杂合子(α/α-),缺失两个α基因,间或有轻度贫血,我国主要是α0地贫杂合子。
轻型α地贫患者之间婚配,生育子女中可有1/4机会为HbBart水肿胎儿综合征。
④静止型α地中海贫血:仅缺失一个α基因,为α+地贫杂合子(-α/αα),无症状。
静止型α地贫与轻型地中海贫血个体婚配,可有1/4机会生育HbH病患儿。
2)α地中海贫血的分子基础:从基因缺陷程度来区分,可把α地贫分为缺失型和非缺失型(点突变)。
①基因缺失:可分为α+和α0地贫两种。α+地贫有两种基因型:左侧缺失(leftward deletion),缺失一个包括α2基因在内的DNA片段;②右侧缺失(right ward deletion), 缺失范围包括α2基因3’端和α1基因的5’端,结果形成了由α1的3’端和α2的5’端构成的融合基因。其发生机理是类α基因发生不等交换的结果。α0地贫基因缺失范围差别很大(图4-15)。

图4-15 类α基因簇缺失类型
②非缺失型(点突变:(类型见表4-1)。
(2)β地中海贫血:β地中海贫血(β梩halassemia,简称β地贫),是由于β珠蛋白基因的缺失或缺陷使β珠蛋白链(简称β链)的合成受到抑制而引起的溶血性贫血。完全不能合成β链者称β0地贫;能部分合成β链者(约为正常的5%-30%)称β+地贫。此外,还有δβ地贫。它们可以有不同的组合,即β0地贫纯合子(β0β0)、β0地贫双重杂合子(β0/β+)、β0地贫杂合子(β0βA)、β+地贫纯合子(β+/β+)和β+地贫杂合子(β+/βA)。β地贫在我国南方较常见。
1)临床分类:大致可有4种主要类型。
①重型β地中海贫血:患者是β+地贫、β0地贫或δβ0地贫的纯合子(其基因型分别为β+/β+、β0/β0和δβ0/δβ0)或是β+和β0地贫的双重杂合子(基因型为β0/β+)。这些患者的β链几乎不能合成,或合成量很少,以致无HbA或量很低,γ链的合成相对增加,使HbFt GbA2比率升高。由于HbF较HbA的氧亲和力高,在组织中不易释放出氧,所有β地贫患者有组织缺氧症状。组织缺氧促使红细胞生成素大量分泌,刺激骨髓的造血功能,使红骨髓大量增生,骨质受锓蚀致骨质疏松,可出现“地中海贫血面容”(头颅大,额顶及枕部隆起,鼻梁塌陷,上颌及牙齿前突,眼距宽,眼睑浮肿)。由于β链合成受抑制,过剩的游离α链形成α链包涵体,引起溶血性贫血,靠输血维持生命。
表4-1 点突变引起的地中海贫血
| 分子缺陷类型 |
| 1、生成无功能或稳定性降低的 ①无义突变 a116GAG---TAG,(G---T) ②移码突变 a130/31(--4bp) ③终止密码突变 142TAA---CAA(T---C) 形成Hb Constant Spring ④起始密码突变 a2ATG---ACG(T--C) 2、RNA加工突变 ①剪接改变 IVSI(GGTGAGGCT---GGCT) ②Poly(A)信号缺陷 AATAAAA ---AATAAG 3、产生不稳定H a2125CTG(亮)CCG(脯) 生成Hb Quong Sze |
②轻型β地中海贫血:患者是β+地贫、β0地贫或δβ0地贫的杂合子,基因型分别为β+/βA、β0/β+和δβ0/βA。这类患者由于还能合成相当量的β链,所以症状较轻,贫血不明显或轻度贫血。本病特点是HbA2升高(可达4%-8%)或(和)HbF升高。
③中间型β地中海贫血:患者通常是某些β地贫变异型的纯合子,如β+地贫(高F)/β+地贫(高F)或两种不同变异型地贫的双重杂合子,如β+,地贫/δβ+地贫。其症状介于重型和轻型之间,故称为中间型β地中海贫血。
④遗传胎儿血红蛋白持续增多症:患者是由于β基因簇中某些DNA片段的缺失或者点突变,使δ和β链合成受抑制,而γ链的合成明显增加,使成人红细胞内HbF含量持续增多,故称为遗传性胎儿血红蛋白持续增多症(hereditarypersistance of fetal hemoglobin,HPFH).HPFH的特点是HbF的成年人仍持续较高水平,无明显的临床症状。
2)β地中海贫血的分子基因:β地中海贫血迄今已发现100多种突变类型,其中10多种为缺失型,其余均为点突变。我国已报道17种点突变。
点突变:绝大多数β地中海贫血是由于β基因发生点突变所致,突变涉及基因内及旁侧表达顺序的各个环节。主要类型有4种。
a.编码区的无义突变、移码突变和起始密码突变:使生成的mRNA稳定性降低或形成无功能的mRNA,从而不能合成正常的β珠蛋白链,多数产生β0地贫,少数为β+地贫。例如无义突变密码子17(A→G)、43(G→T)都产生β0地贫;称码突变41/42(-TCTT),71/71(+A)和β0地贫;以及起始密码子突变ATG→AGG导致的β0都属这类。见于中国人。
b.非编码区IVS-1和IVS-2突变:影响前mRNA剪接等加工过程不能准确进行,形成异常的mRNA,导致β0或β+地贫。例如IVS-1的1位G→T为RNA拼接处改变;中国人中常见的IVS-2654,为内含子中由于碱基置换形成了一个新的裂解信号,影响正常位点的剪接,产生异常mRNA;还有一种是内含子中剪接位点的通用顺序上的同义突变,从而激活内含子或外显子中隐蔽裂解位点(cryptic splicihng site,CSS)如IVS-15(G→C)。CSS即DNA的一段顺序在点突变后可以形成剪切识别顺序(CCTATTGGT)的第7个碱基G如变为A,则产生新的切点,即CCTATTAG↓T)。
c.影响转录的突变:这类突变主要集中于起始位点上游的启动子TATA框,使转录效率降低,mRNA生成量减少而产生β+地贫。中国人的-29A→G,-28A→G都属于这类。
d.RNA裂解部位缺陷:这类突变是由于异常的RNA加帽部位和多聚腺苷酸化信号的突变,从而影响RNA转录而不能准确裂解,产生不稳定的mRNA,使正常β链生成量减少,导致β+地贫。例如在mRNA加帽部位发生A→C颠换,引起β+地贫(亚洲人。)又如,多聚腺苷酸化信号AATAAA→AACAAA,引起β+地贫。
e.编码区的外显子突变引起剪接作用的改变:这类突变是由于编码区的单碱基突变(错义突变或同义突变)激活邻近的隐蔽裂解信号,影响IVS正常位点的剪接,产生异常的mRNA。如东南亚常见HbE,是一种轻型的β地贫,其原因是当β链26位密码子发生G→A错义突变时,其相邻的裂解信号被激活,生成异常mRNA,产生HbE(图4-16)。
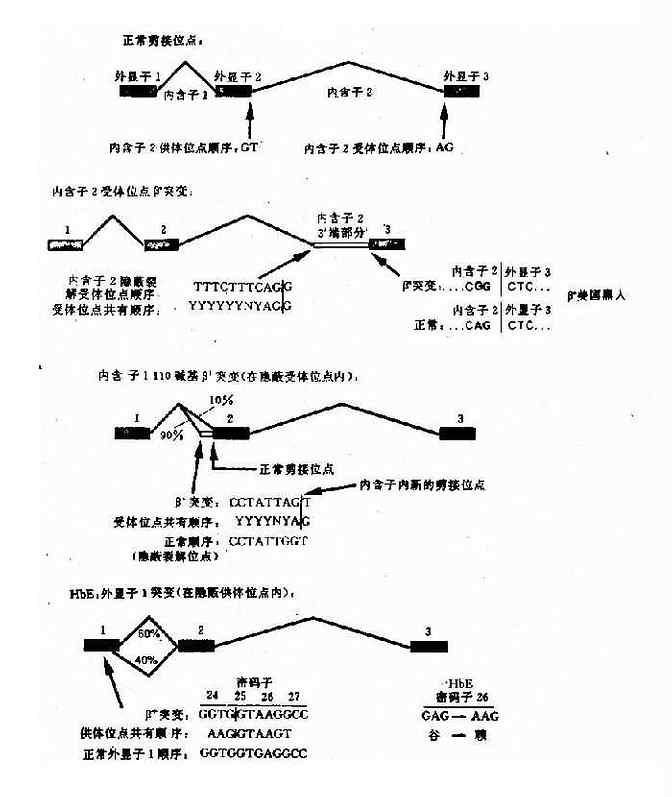
图4-16 干扰正常β珠蛋白剪接的突变举例
HbE:密码子26(G-→A)
谷→赖
GAG→AAG(QAJ)(错义突变)
β+(HbA 60%) βE(HbE 40%)
3)类β基因缺失:①按类β珠蛋白基因簇缺失长短大致可分为4种,即β0、δβ、γδ地中海贫血及遗传性胎儿血红蛋白持续增多症;②单纯由于β0基因缺失引起的β地中海贫血罕见;③融合基因,如HbLeproe,是类β基因缺失7kb导致δβ融合基因,形成β0地贫。
二、免疫缺陷病
免疫缺陷病(immunodeficiency)是指免疫系统功能障碍引起的一类疾病,就其原因可分为遗传性(原发性)与继发性两大类。根据参与免疫反应的细胞又可分为B细胞和T细胞以及联合免疫缺陷病。本节主要阐述具有免疫功能的各种蛋白质遗传性缺陷引起的免疫缺陷病。主要有下列几种:
1.无丙球蛋白血症无丙球蛋白血症(agammaglobulinemia)分为两型:
(1)Bruton型:此型呈X连锁隐性遗传。血中B细胞缺如,导致血中IgA、IgG和IgM完全或基本缺如。而T细胞不受影响。患儿反复发生严重的细菌感染,但对病毒或真菌并无易感性,细胞免疫机制尚保存,对移植物有排斥反应。另有一型X连锁的低丙球蛋白血症(hypogammaglobulinemia)患者,除易发感染外,尚伴有生长激素缺乏所引起的生长发育迟缓,身材矮小,性发育延迟等症状(Fleischer 综合征)。
(2)瑞士型:瑞士型(Swiss type)呈常染色体隐性遗传。血中B细胞及T细胞均缺如,胸腺发育不全,对细菌、真菌及病毒抵抗力均低,对移植物无排斥能力。血中IgA、IgG和IgM均缺如或严重低下。预后较Bruton型更差,常于1岁内死亡。本型有异质性,也发现有X连锁型存在。
2.严重联合免疫缺陷病严重联合免疫缺陷病(severecombined immunodeficiency)是指B细胞和T细胞均有缺陷的一类免疫缺陷病。一般分为4类,瑞士型无丙球蛋白血症属其中一类,其它3类为:X连锁淋巴细胞减少性低丙球蛋白血症、网状细胞发育不全和腺苷脱氨酶缺乏症。
3.选择性免疫球蛋白缺乏症由于检测免疫球蛋白技术的改进,现在已有可能降低丙球蛋白血症按其主要缺乏的免疫球蛋白类型分为8类(表4-2)。
表4-2选择性免疫蛋白缺乏症分型
| IgA缺乏症 选择性IgA缺乏症 共济失调-毛细血管扩张症 Nezelof综合症 慢性皮肤粘膜念珠菌病 SIgA(分泌型)缺乏症 IgA缺乏症 IgA缺乏症 选择性IgM缺乏症 Wiskott-Aldrich综合征 IgE缺乏症 IgA-IgM缺乏症 IgA-IgG缺乏症 L链缺乏症 |
选择性IgA缺乏症为最常见类型。本病主要表现为上呼吸道感染、肺炎、支气管炎,其次为肠炎、自身免疫性疾病(类风湿关节炎、系统性红斑狼疮)等。
4.补体成分缺乏症补体(cpmplement)是一个比较复杂的系统,包括12种成份,还有接近补体性质解裂素系统P因子(解裂素)、B因子和D因子,此外,还有几种调节蛋白B如Ci抑制因子(Ci INH)、C3b激活因子(3bINA)、βIH因子等。
基因突变可以导致补体合成障碍,引起补体缺乏症。已报告过的补体缺乏症至少有14种,包括:Ci INH,C1q,C1r,C2,C3,C3b灭活剂,C4,C5,C6,C7,C8,C9缺乏症及C5功能异常。这类疾病的主要表现是反复感染、皮下和粘膜水肿及自身免疫性疾患。
三、膜蛋白病
细胞膜是物质、能量、信息总的传递和变换的场所,具有广泛的生理功能,其中有许多功能是与膜蛋白息息相关的。按其功能可分为下列几类。
1.膜骨架蛋白病以红细胞为例。红细胞膜是由双层脂质组成,外层主要为胆碱磷脂,内层主要为氨基磷脂,横跨双脂层及双脂层下有多种蛋白质*,横跨双脂层的蛋白质称为整合膜蛋白或跨膜蛋白(transmembrane proteins)(图4-17),其中有:①血型糖蛋白A、B、C(glycophorin,GP-A,B、C),有抗原和受体功能;②蛋白3为主要的阴离子转运蛋白,作为运输渠道。
* 膜蛋白的命名沿用电泳速率快慢依次为蛋白1-8,后来又分为亚带蛋白,如蛋白4有蛋白4.1、4.9等,当这些蛋白质的性质或功能明确后,有的名称又有改变,如蛋白2.1又称为锚蛋白等。
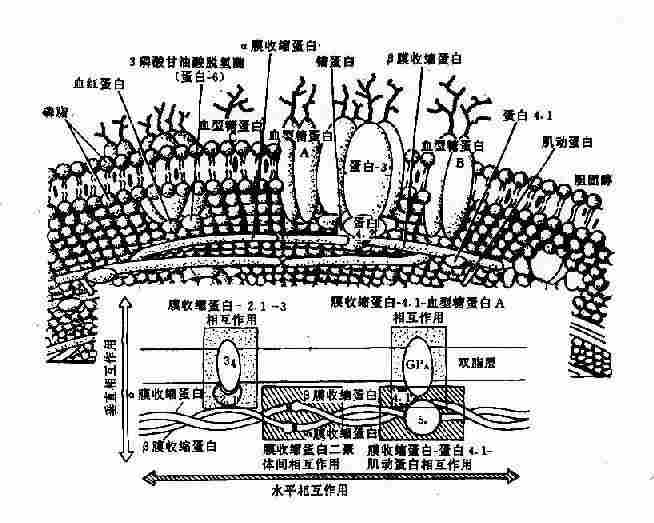
图4-17 红细胞膜的结构示意图
脂质内层与胞质接触部份的蛋白质称外周膜蛋白(peripheral membrane protein),其中有:①膜收缩蛋白(spectrin,Sp)是主要成分,由两种肽链组成,即α-Sp(240kd)与β-Sp(220kd)形成的二聚体(异常二聚体,heterodimer);②膜动蛋白(actin,蛋白5),呈短丝状,连接Sp的四聚体;③蛋白4.1其功能为促进Sp与膜动蛋白结合,并与GP-C连接,亦即将Sp固定在膜上;④蛋白4.2,它与阴离子通道、锚蛋白及蛋白4.1结合;⑤蛋白4.9,与Sp-Sp四聚体结合,再与Sp和蛋白4.1结合,构成红细胞膜骨架(membrane skeleton)的主体(图4-17);⑥锚蛋白(ankyrin,蛋白2.1),其作用似乎是连接β-Sp与蛋白3,使膜骨架蛋白固定在脂质内层,起“锚”的作用。其它尚有一些膜蛋白,其作用在探讨中。膜骨架的形成是维持红细胞双凹形结构、膜的可变形性和完整性的基础。基因突变导致这些膜蛋白结构和功能的改变,是一类遗传性溶血性贫血的根本原因。
(1)球形细胞增多平:球形细胞增多症(spherocytosis)是以球形红细胞增多为特点的溶血性疾病。患者呈慢性中度贫血、黄疸、脾大,大多数红细胞呈球形,脆性明显增高。本病多数呈常染色体显性遗传。从分子水平看,有遗传异质性,有Sp轻至中度缺乏、锚蛋白缺乏、蛋白4.2缺乏等病因。
(2)椭圆形细胞增多症:正常外周血中有1%-14%的椭圆形红细胞,而遗传性椭圆形红细胞增多症时,可增至50%-90%,出现溶血、贫血、黄疸、脾大等症状,红细胞脆性增高。多呈常染色体不完全显性遗传,其遗传基础也有异质性,可因Sp二聚体结合障碍、蛋白4.1异常或糖蛋白缺乏引起。
(3)热异形细胞增多症:热异形细胞增多症(pyropoikilocytosis,HPP)的特点是外周血红细胞对热不稳定,在加温至46℃时即出现异红细胞乃至红细胞碎片(正常要49℃才出现)。患者表现严重溶血。呈常染色体隐性遗传。Sp严重缺乏是本病的基本原因。
目前发现的几十种膜蛋白基因突变与上述疾病有关,但某种突变与这三种疾病间并没有发现必然的联系,但Sp突变仍占多数。
2.膜转运蛋白病有些物质(如氨基酸、葡萄糖)通过细胞膜需要借助于分子导体(膜蛋白)进行易化扩散(faciliteddiffusion)。基因突变可导致膜蛋白质或量的改变,影响某些物质通过细胞膜的转运,由此产生的一类疾病称为转运病(transport disease)。例如胱氨酸尿症就是由于肾近曲小管和胃肠上皮细胞不能转运胱、赖、精和鸟氨酸4种氨基酸。其中特别是胱氨酸解度低,在尿中浓度超过30mg%时,即易产生胱氨酸结石。因此,尿路结石、感染、绞痛是本病的主要临床表现。目前已知的转运病有十多种。
3.肌膜蛋白病肌膜蛋白病以附着于肌膜(sarcolemma)上的抗肌萎缩蛋白(dystrophin,又称肌营养不良蛋白)遗传缺陷为代表的假肥大型肌营养不良症(又称Duchenne型肌营养不良症,DMD)和表现较累的良性假肥大型肌营养不良症(又称Becker型肌营养不良症,BMD)加以讨论。
DMD是一种严重致残致死性X连锁隐性遗传病,发病率约为1/3500活男婴。其临床表现以肌肉的进行性萎缩和无力为特征。患者多在3-5岁发病,肌肉无力,走路困难呈鸭步,大部部分患者有腓肠肌假性肥大现象和不同程度的心肌损害,约30%伴有智力障碍。肌纤维萎缩变性,被脂肪及结缔组织替代。血清磷酸肌酸激酶等活性升高。多在12岁左右便不能行走,一般在20岁前死亡。BMD患者病情较轻,临床表现与DMD相似,在5-25岁发病,病程发展较缓慢,存活期较长,有活至63岁者。现在已证实DMD和BMD是同一基因发生不同突变的后果。
人群中DMD病例约2/3有家族史,呈X连锁隐性遗传,有约1/3病例为散发,是由基因新突变造成。DMD基因定位在Xp21,全长约2300kb,为迄今人类认识的最大基因。此基因至少含有79个外显子,内含子长度差异很大,可从1kb-180kb。cDNA全长13,974bp,编码3685个氨基酸,分子量为427kd。根据电脑提示该基因产物为一棒状结构的细胞骨架蛋白,可分为4个功能区(图4-18)。
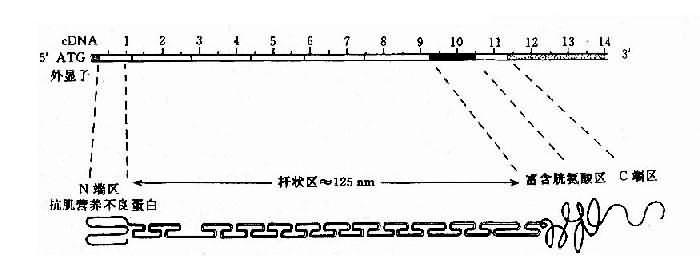
图4-18 DMD基因及其编码蛋白质的结构示意图
目前已知的DMD基因突变主要为缺失型,约占病例的50%-60%;重复(duplication)次之,约占6%,有两个缺失热区:即5’端的第4-21外显子(占缺失的20%);另一为第45-52外显子(占54%-60%)。内含子44约160-180kb,断裂频率最高,缺失导致移码突变者,多数会引起DMD,未导致移码者多为BMD。无论缺失或重复,都有可能使突变区域的两个残端形成连接片段,这对于家系分析、携带者检出及产前基因诊断都极有意义。
DMD基因上还存在一些短串联重复顺序(short tandem repeats,STR),已知有13个STR。DMD的STR多表现为两个核苷酸亚单元的串联重复,如CA串联(Ca repeats)。重复频数在不同个体差异很大,杂合子频率达25%-93%,为非缺失型DMD的产前诊断、携带者检出提供了一种有效的遗传标记。
目前尚无有效疗法,唯一有效的预防途径是对高风险胎儿进行产前诊断(参阅第十三章)。
四、凝血及抗凝血因子缺乏症
凝血是一复杂的生理过程,包括多种凝血因子及抗凝血因子参与。遗传性凝血障碍包括低凝和高凝两种情况。前者是由于基因突变致凝血因子活性降低,后者是由于抗凝血因子活性缺乏。
1.凝血因子缺乏症
除Ca2+与凝血活素外,所有凝血因子都有遗传性缺乏的报告,其中血友病,特别是甲型血友病较为多见。现主要就血友病及与其有关的血管性假血友病加以介绍。
血友病(hemophilia)除甲、乙、丙三型外,加上后来又发现的一种vWF因子(vonwillibrand factor)缺乏的血管性假血友病,构成血友病的四种类型。1986-1989年全国24省、市,37个地区作血友病患病率的调查,调查人数16866 654人,总患病率2.73/10万(男性5.21/10万,女性0.06/10万),与欧美相比较低,四型的构成比是:甲型79.8%,乙型14.1%,丙型2.8%,血管性假血友病3.3%。
(1)甲型血友病:甲型血友病(hemophilia A)又名抗血友病球蛋白(antihemophilicglobulin,AHG)缺乏症或第Ⅷ因子缺乏症。主要表现为出血倾向,其出血特点为:①缓慢持续渗血;②多发生于轻微创伤之后;③出血部位广泛,常反复发生,可形成血肿,并节变形,死因多为颅内出血。
现知,Ⅷ因子由三种成分组成:①FⅧ:C(AHG);②FⅧ:Ag(Ⅷ因子相关抗原);③vWF因子。甲型血友病为AHG遗传性缺乏所致。
本病为X连锁隐性遗传,基因定位于Xq28,基因跨度超过186kb,由26个外显子(占9kb)及25个内含子(占177kb)组成,编码2351个氨基酸,已发现缺失型(包括错义、无义及移码突变等)共46种以上。杂合子的鉴定对开展遗传咨询很重要。过去多通过测定血浆AHG水平或用ⅧR:Ag/AHG比值来检出杂合子,现已能采取分子遗传学手段,特别是已成功地应用DNA印迹杂交、PCR技术等于产前诊断,这对防止重型患儿出生,十分有效。
治疗可使用各种AHG制剂,但需长期使用,正在研究的基因治疗将会是本病的根治方法。
(2)乙型血友病:乙型血友病(hemophilia B)又名血浆凝血活酶成分(PTC)缺乏症或第Ⅸ因子缺乏症。此型临床表现酷似甲型,但发病率较低,遗传方式亦为Ⅹ连锁隐性遗传,由于杂合子Ⅸ因子活性仅为正常1/3,某些杂合子可出现症状,故女性病人较甲型多见。
人类第Ⅸ因子基因已定位Xq27.1,基因总长度为34kb左右,由8个外显子组成。已鉴定出的突变有100种之多(部分缺失及全缺失者30种,其余为各种类型点突变)。我国王宁波等报告了重庆发现的5种点突变(外显子2和5)。上海曾溢滔等发现1例1-3内含子5kb片段缺失。在这些突变中发现几例启动子突变,临床表现为儿童期有严重出血倾向,但到青春期后自发出血减轻。后证明,雄性类固醇可诱导启动子产生Ⅸ因子,这也表明临床症状轻重与突变性质可以有一定关系。
本型亦多采用输血浆或浓缩血浆制剂治疗,将第Ⅸ因子活性提高到25%以上即有疗效。产前诊断是防止本病患儿出生的有效方法。近年,我国薜京伦等在Ⅸ因子缺乏症的基因治疗方面取得了进展,有望在临床取得长期稳定疗效。
(3)丙型血友病:丙型血友病(hemophilia C)又名血浆凝血活酶前质(PTA)缺乏症(plasma thromboplastic antecedent deficiency)或第Ⅸ因子缺乏症。此型症状较甲、乙型轻。本病种族倾向明显,多见于土耳其南部犹太人后裔。遗传方式属染色体隐性遗传。现知,基因定位于15q11,基因长度为23kb,由15个外显子组成,编码625个氨基酸,但仅第11-15外显子编码羧基端具有凝血功能为Ⅺ因子主要成分。已发现3种点突变。本病纯合子的Ⅺ因子活性在20%以下,杂合子为30%-65%。多数严重缺乏者用小剂量正常或浓缩血浆治疗即显效。
(4)血管性假血友病:血管性假血友病(von Willebrand disease)是一种较多见的与第Ⅷ因子有关的遗传性凝血障碍。与本病有关的von Willebrand因子(v WF)是一大分子量的糖蛋白。基因定位于12pter-p12,vWF基因长度为180kb,有52个外显子,mRNA总长度为9kb左右编码2813个氨基酸。vWF蛋白由血管内皮细胞及巨噬细胞分泌,它在血中不仅作为Ⅷ因子载体,而且可增强Ⅷ稳定性,故vWF因子缺乏往往伴有Ⅷ因子活性降低。此外,血小板α颗粒中含vWF,故也参与血小板聚集,在凝血中发挥作用。本病患者有明显的出血倾向。血中AGH活性降低,但不如甲型血友病严重,从基因水平已发现20多种突变型。
由于对本病基因有足够的了解,已可通过RFLP连锁分析或PCR法对本病进行产前诊断。
此外,由于遗传性血小板缺乏或功能障碍和纤维蛋白原的遗传性缺陷都可造成低凝状态,在此不多赘述。
2.抗凝血因子缺乏症
(1)遗传性抗凝血酶Ⅲ缺乏症:抗凝血酶Ⅲ(antithrombinⅢ,ATⅢ)对凝血酶Xa有抑制作用,肝素能加速其对凝血酶的抑制。其次,ATⅢ还有抑制Ⅸ、Ⅺ及Ⅻ的功能。
遗传性抗凝血酶Ⅲ缺乏症(hereditary antithrombin Ⅲ deficiency)的临床表现为容易发生血栓形成(主要部位为髂静脉)及肺体塞。
本病为常染色体显性遗传。发病率在不同种族有显著差异。欧美白种人中可高1:2000-5000。我国也已有个例报告。现知,ATⅢ基因定位1q23,基因长16kb,由7个外显子组成,编码432个氨基酸,至少已发现20种以上的突变类型,表现出不同的功能缺陷。
(2)遗传性蛋白C系统异常:遗传性蛋白C系统由蛋白C(PC)、蛋白S(PS)、血栓调节蛋白(thrombomdorlin,TM)及活性蛋白C抑制物(APC1)组成,它们之间的关系见图4-19。活化的蛋白C有抑制Va、Ⅻa及促进纤维蛋白溶解的作用。蛋白C系统缺乏症有3种:
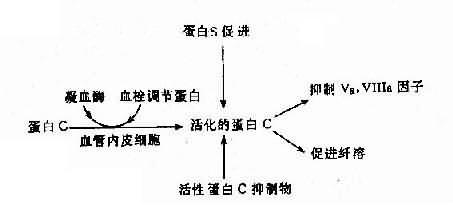
图4-19 蛋白C、蛋白S及活性蛋白C抑制物的相互关系
1)蛋白C缺乏症:主要症状是静脉血栓形成,常见于深部静脉。本病发生率估计为1:16000,呈常染色体显性(或不完全显性)遗传,纯合子严重,表现为出血性皮肤坏死、弥漫性血管内凝血(DIC)和血栓。杂合子多数在青壮年发病。蛋白C基因定位2号染色体,基因长12kb,由8个外显子组成,已鉴定出若干种缺失型及点突变病例,也有涉及mRAN加工缺陷者。
2)蛋白S缺乏症:蛋白S的作用是促进活化蛋白C(APC)结合于磷脂,加速APC灭活Va因子。本病亦属常染色体显性(或不完全显性)遗传。基因定位在3号染色体,其总长度为45kb。上海也已报告2例PS缺乏症。
3)先天性活化蛋白C抑制缺乏症(congenitaldeficiency of activated protein Cinhibitor)。
其它还有遗传性纤溶系统异常,包括①先天性异常纤溶酶原血症;②先天性纤溶酶原激活物释放异常;③遗传性纤溶酶原激活抑制物增多症;④血块异常所致先天性纤溶减弱等,均表现出高凝状态的各种症状。
五、受体蛋白病
受体是存在于细胞膜上、胞质中或核内的一类具有特殊功能的蛋白质。现已证明具有调节生理功能和特异性受体达30种以上。其中包括多肽激素受体、固醇类激素受体以及神经递质、前列腺素、免疫性因子、脂蛋白等受体。由于受体的本制是蛋白质,不言而喻,基因突变也可导致受体蛋白质和量的改变。由于受体蛋白遗传性缺陷引起的一类疾病,称为受体病(receptor diseae)。
受体病有获得性与遗传性之分。遗传性受体病研究得较多的有下列4种:
1.家族性高胆固醇血症家族性高胆固醇血症(familialhypercholesterolemia,FH)的临床表现是:出生时即存在高胆固醇血症,增高的胆固醇主要为低密度脂蛋白胆固醇()LDL-C)和β极低密度脂蛋白(β-LDLC),黄色瘤(xanthoma)即增高的胆固醇在组织广泛沉积。其最严重的后果是早年发生动脉粥样硬化。纯合子在5-30岁即表现心绞痛和心肌梗塞症状,可骤死。杂合子发生冠心病稍迟且发生率较低。
本病为常染色体显性遗传病。在人群中,杂合子发生率为1:500,杂合子临床表现较轻,故属不完全显性遗传,外显率高(90%-100%)。
本病的主要病因是LDL受体(LDLR)的遗传性缺乏。血浆中的LDL和β-VLDL与LDL受体结合后,内吞入细胞,在溶酶体中LDL与LDLR分离,LDLR回到细胞表面重新利用,LDL则在胆固醇酯酶作用下,释出胆固醇供细胞利用。在这一过程中,细胞表面有功能的LDLR数量决定了血浆中LDL及β-VLDL的浓度。现知LDLR的编码基因位于19p1.3-p13.3,总长度45kb,由18个外显子组成。已发现FH患者有数十种突变,包括缺失型突变(主要的)、错义突变、无义突变、移码突变及整码突变。表现为LDLR功能异常有4种类型:①细胞膜上完全无LDLR;②LDLR明显减少;③LDLR无功能不能与LDL结合;④LDLR与LDL结合后不能内吞,可能是由于合成受体前后有加工缺陷。
2.睾丸女性化综合征睾丸女性化综合征(testicularferminization syndrome)又称雄性素全不敏感综合征(complete androgeninsensitivity syndrome,CAIS),为最常见男性假两性畸形。患者核型为46,XY,有睾丸,能分泌雄性素,但缺乏雄性素受体,雄性素不能发生效应(图4-20)。
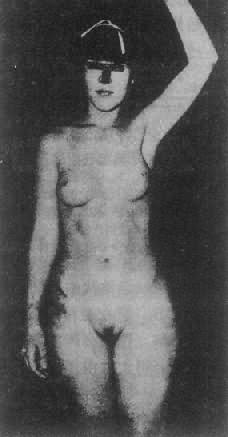
图4-20睾丸女性化综合征患者(核型46,XY)
第二节 基因突变致酶活性异常
由于基因突变导致酶活性降低或增高所引起的疾病称为遗传性酶病(hereditary enzymopathy)。遗传性酶病与分子病的区别在于后者引起机体功能障碍是蛋白质分子变异的直接后果;而前者则由于合成酶蛋白结构异常或调控系统突变后导致酶蛋白合成数量减少,通过酶的催化作用间接导致代谢紊乱所引起的机体功能障碍。基因突变导致酶的遗传变异可表现为酶活性降低、酶活性正常(同义突变或突变部位不影响酶活性中心)及酶活性增高。绝大多数遗传性酶病是由于酶活性降低引起的,仅少数表现为酶活性增高。
据估计,人类的酶有10 000种左右,但目前已明确的酶仅200多种,只占酶总数的2%左右。遗传方式以常染色体隐性遗传为多见,常染色体显性和X连锁隐性遗传者较少。杂合子所产生酶量往往等于正常纯合子和突变基因纯合子所含酶的半量,这种现象称为基因的剂量效应(gene dosage effect)。
一、酶活性降低引起的遗传性酶病
1.酶活性降低的原因基因突变引起酶活性降低的可能原因包括:①结构基因突变;其结果致使酶动力学特性改变(表现为酶与底物亲和力降低,与抑制物的亲和力增高)和酶的稳定性降低(表现为酶降解速率加快);②调节基因突变:酶合成速率减慢;③影响翻译后修饰和加工。
图表-21 典型代谢途径示意图
S1:底物;S2、S3:中间产物; P:终产物;E1-2、E2-3:不同代谢环节的酶;
E3-P:形成终产物的酶;E6-7:旁路代谢的酶;S6、S7:
旁路代谢产物;---旁路代谢途径;---反馈抑制
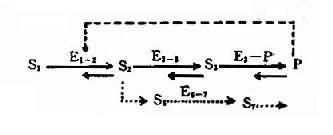
2,酶活性降低的发病机理人类体内一般代射过程如图4-21所示:代谢底物(S1)进入细胞后,底物(S1)在一系列酶(E+1-2。E2-3,E3-P)催化下经过一系列中间产物(S2、S3),最后形成代谢终产物(P),同时代谢产物(P)可对一定的酶(如E1-2酶)起反馈抑制作用。另外,在正常代谢过程中还可能存在代谢旁路(S2→S6→S7……)。当控制酶蛋白合成的基因发生突变时,正常代谢受阻,可能通过不同发病环节引起不良后果。
(1)酶缺乏致代谢中间产物堆积和排出引起的疾病:当E2-3缺乏时,中间产物S2不能转变为S3,它在血和尿中的浓度增加,如果中间产物是无毒的,可由肾排出或通过其它方式降解,则不会严重危害人体。如尿黑酸尿平患者,因酪氨酸代谢中间产物(尿黑酸)堆积,可大量由尿排出,对人体无影响。但如果中间产物具有毒性,则将会引起症状。
半乳糖血症可作为实例,半乳糖血症(galactosemia)表现为婴儿哺乳后呕吐、腹泻,对乳类不能耐受,继而出现肝硬化、白内障、智力发育不全等症状。乳类含有乳糖,它经消化道乳糖酶分解产生葡萄糖及半乳糖,半乳糖通过一系列酶促反应产生葡萄糖而被组织利用(图4-22)。
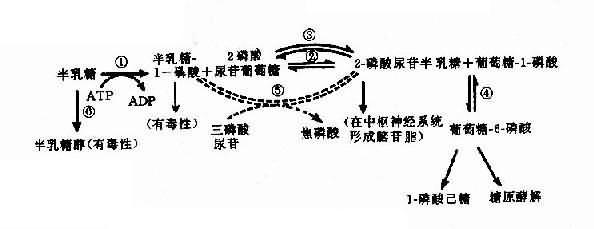
图4-22 半乳糖代谢途径(虚线表示年长以后才发展起来的代偿途径)
①半乳糖激酶;②半乳糖-1-磷酸尿苷酰转移酶;③半乳糖尿苷-2-磷酸-4-异构酶;④磷酸葡萄糖变位酶;⑤-磷酸尿苷半乳糖(或葡萄糖)焦磷酸化酶;⑥醛糖还原酶
典型的半乳糖血症患者由于半乳糖-1-磷酸尿苷酰转移酶(galactose-1-phosphate uridy1 transferase,简称转移酶)缺乏(图4-22②),致使半乳糖-1-磷酸(Gal-1-P)及半乳糖积聚在血中,部分随尿排出。Gal-1-P在肝的积聚可引起肝功能损害,甚至肝硬化;在脑的积聚引起智力障碍;血中半乳糖升高可使葡萄糖释出减少,出现低血糖症。半乳糖在醛糖还原酶作用下产生半乳糖,能改变晶状的渗透压,使水分进入,影响晶状体代谢而致白内障。
患者都是隐性纯合子(gg),杂合子表型正常,转移酶活性约为50%,活性低于10%可出现典型症状。
另一类半乳糖血症为半乳糖激酶(galactokinase)缺乏所引起,症状较轻,主要表现为青年型白内障,血中半乳糖增高,但无肝及脑损害。
转移酶基因(GALL)定位于9p13,半乳糖激酶基因(GALK)定位于17q21-q22。两病均为常染色体隐性遗传。
(2)酶缺乏致代谢底物堆积引起的疾病:当一系列生化反应可逆时,一处的阻断常导致代谢底物(S1)贮积。贮积的物质如果溶解度高,则在血和尿中浓度增高;若溶解度低时,则在组织中贮积引起疾病。
糖原贮积症(glycogenstorage liseaee,GSD)是一组由糖原合成和降解酶缺陷引起的疾病,至少有12种类型。糖原贮积症主要累及肝或肌肉,但有的也可伴有心、肾和神经系统的损害。不同类型之间其严重性和预后都不全相同。例如,von Girke病(Ⅰ型)症状非常严重,而Hers病(Ⅵ型)较轻。此类疾病发病机理可用von Gierke病为例说明。本病是由于肝内葡萄糖6-磷酸酶(glucose-6-phosphatase,G6Pase)缺乏引起。肝糖原在一系列酶的作用下生成葡萄糖,这个反应的各个步骤都是可逆的,其主要步骤如下:
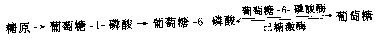
患者由于G6Pase缺乏,所以G6P不能转变为葡萄糖供组织利用,通过可逆反应而合成过多的肝糖原,引起患儿肝肿大。当不进食时极易发生低血糖。由于动用脂肪可以出现酮血症。G6P通过无氧酵解,生成大量乳酸,导致酸中毒。所以患者的肝大伴低血糖,发育不良,消瘦,身体矮小,常有出血倾向。肝活检见糖原含量增加。
本病呈染色体隐性遗传。患者G6P酶完全缺乏,多数患者父母表型正常,但G6P酶活性为中间值。同胞中可出现患者。
(3)酶缺乏致代谢终产物缺乏引起的疾病:这是一类由E2-3酶缺乏,致所有产物(包括终产物P)缺乏引起的疾病.白化病就是实例.白化病(albinism)分全身型及局部型.全身型常见,患者皮肤呈白色,毛发银白或淡黄色,虹膜及瞳孔呈淡红色,视网膜无色素,羞明,眼球震颤等,本病发病率约1/10000-1/20000,呈常染色体隐性遗传.
正常人黑色素由黑素细胞合成,这些细胞中有特殊的细胞器-黑素小体(melanosome),其中有含铜的酚氧化酶,即酪氨酸酶(tyrosinase),它可将酪氨酸转变成黑色素(图4-23).白化病人有黑素细胞,但酪氨酸酶缺乏,使黑色素不能形成,病人因缺黑色素而白化.白化病存在遗传异质性.已知白化病至少有7种不同类型.酪氨酸酶基因(TYR)定位于11q14-q22.
属于此类的例子尚有遗传性甲状腺肿,它是由于偶联酶、脱碘酶或碘过氧化酶缺乏导致甲状腺素生成减少引起的,患者身材矮小,智力低下,面貌丑陋.
(4)酶缺乏致旁路产物增多引起的疾病:当酶的缺乏导致主要代谢途径受阻断时,过量的前体物S2通过另一旁路代谢引起某些副产物的堆积.如果增多的旁路产物是无毒的,则可排出体外不致危害人体,但如果旁路产物或其分解产物有毒,则可危害机体引起疾病.
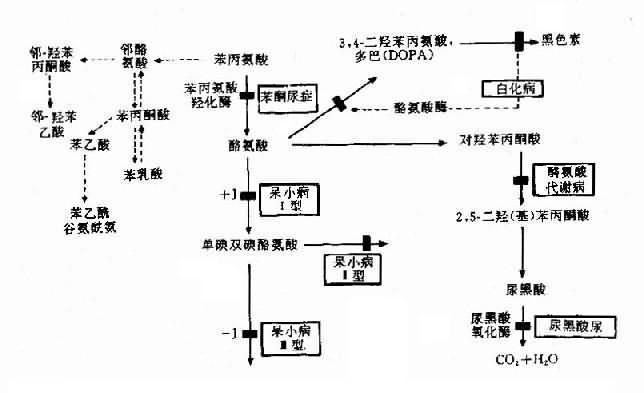
图4-23 苯现氨酸代谢及有关的遗传性酶病的发病机理
苯酮尿症(phenylketouria,PKU)可作为实例。本病以智能发育不全为主要特征,呈常染色体隐性遗传,群体发病率约1/16000,由苯丙氨酸羟化酶(phenylalaninehydroxylase,PAH)遗传性缺乏引起。现已知苯丙氨酸羟化酶基因定位于12q24.1,此基因全长约90kb,含13个外显子,在中国人中已发现10余种点突变,这是造成酶活性缺乏的原因。
典型PKU患儿出生时,外貌正常,约至3-4个月时,渐出现智能发育不全,患儿步伐小,姿似猿猴,肌张力增高,易激动,甚至惊厥,90%以上患者毛发发黄,肤白,甚至虹膜呈黄色(白种人呈蓝色)。此外,患儿尿和汗有一种特殊的腐臭。
正常人苯丙氨酸在体内主要通过PAH的作用转变成为酪氨酸,继而生成黑色素(图4-23)。PAH主要存在于肝中,它的作用需要辅因子四氢生物蝶啶(BH4),如果PAH缺乏(活性<1/10)可阻断苯丙氨酸转化为酪氨酸,即产生典型的苯酮尿症。酶活性部分缺乏,可导致轻度高苯丙氨酸血症(hyperphenylalaninemia)。
典型PKU患者,由于肝内PAH几乎完全缺如,致血清苯丙氨酸增高(正常人1-3mg%,患者可达50-100mg%)。过多的苯丙氨酸通过转氨酶作用生成苯丙酮酸,再经氧化.脱羧产生苯乳酸.苯乙酸等异常产物,由尿和汗液排出,致患儿的尿和汗呈特殊的腐臭。可能由于旁路产物抑制了脑组织内L-谷氨酸羧酶,使谷氨酸脱羧基生成γ-氨基丁酸减少,而后者对脑细胞的发育及功能起重要作用。也有人认为苯现氨酸和旁路产物可影响5-羟色胺的生成,而影响脑功能。由于正常产物酪氨酸是黑色素前体,所以酪酸不足加之旁路产物可以抑制酷氨酸酶,使患者皮肤毛发及眼血管膜色素减少,因而色泽都较浅。
(5)酶缺乏致反馈抑制减弱引起的疾病:有些代谢过程中,某些代谢产物对整个反应过程具有反馈调节作用。因此,某种酶的遗传性缺陷,使该代谢产物减少,致反馈调节功能失调。自毁容貌综合征可作为实例。处毁容貌综合征亦称Lesch-Nyhan综合征(Lesch-Nyhan syndrome,LNS),是由于遗传性次黄嘌呤鸟嘌呤磷酸核糖转移酶(hypoxanthine-guanine-phosphoribosyltransferase,HGPRT)缺乏所引起。本病的特征是智力发育不全、舞蹈样动作和强迫性自残行为,并伴有高尿酸血症、尿酸尿、血尿、尿道结石和痛风。患者可活至20余岁,多死于感染和肾功能衰竭。
LNS呈X连锁隐性遗传,患者均为男性(半合子)。现知HGPRT定位于Xp26-q27.2,其发病率约为1/380000。
正常时,HGPRT的功能是将次黄嘌呤转变为次黄苷酸(((即IMP,肌苷酸),将鸟嘌呤转为鸟苷酸(GMP),鸟苷酸及腺苷酸可以反馈抑制磷酸核糖焦磷酸(PRRR)合成1-氨基-5-磷酸核糖,从而控制IMP的自发合成速度(图4-24)。这一过程,HGPRT的催化起到反馈抑制作用。当HGPRT缺乏时,这一反馈抑制作用减弱或消失,IMP和GMP生成量大为减少,嘌呤合成速度就大大增快,致使患者体液内的尿酸浓度增高,出现高尿酸血症和尿酸尿。经典型患者无HGPRT活性。酶仅部分缺乏时,患者往往出现痛风而无LNS症状。
图4-24 HGPRT缺乏导致Lesch-Nyhan综合征
5-PR-1-amine:-1氨基-5-磷酸核糖;PRPP-Sym:磷酸核糖焦磷酸全盛酶;虚线:反馈抑制
(6)维生素依赖性遗传病:这是由于基因突变改变了某种酶蛋白,使之与辅酶的相互作用受到损害引起该酶活性降低。这类辅酶多数为维生素,故称为维生素反应性遗传病(vitam-in-responsive hereditary disorders)。例如,在体内,吡多醇-5-磷酸(PLP)是维生素B6的活性形式,它是多种酶蛋白的一种辅酶,因此,某些酶蛋白基因的突变能引起多种遗传性代谢病(图4-25)。例如谷氨酸脱羧酶基因的突变致使酶蛋白和PLP的相互作用受损,将导致γ-氨基丁酸缺乏和痉挛。胱硫醚合成酶基因突变使酶蛋白与PLP的结合部位发生改变而导致胱硫醚尿症。胱硫醚合成酶的缺乏可导致同型胱氨酸尿症。
图4-25 吡多醇-5-磷酸(PLP)作为多种酶的辅酶形式示意图
(7)多种酶缺陷引起的疾病:有些遗传性代谢缺陷,在某一患者不只一种酶缺陷。例如先天性蔗糖不耐受症(congenital sucrose intolerance),患者其异麦芽糖酶和蔗糖酶都缺乏,而枫糖尿症(maple syrup urine disease)病人体内则同时缺乏缬氨酸脱羧酶.亮氨酸脱羧酥酶和异亮氨酸脱羧酶。这种现象可作如下解释:①有缺陷的几种酶均有一条共用的多肽链,当编码这条多肽链的结构基因发生突变时,就会使凡含有这条共同多肽链的各种酶蛋白结构改变而失活;②由于一个酶,致使由这种催化生成的代谢物缺乏,因此由它诱导的各种酶也相应缺乏;③由于一个调控基因发生突变,关闭了邻近几个结构基因,使这些结构基因控制的酶不能产生。
二.酶活性增高引起的遗传性酶病
基因突变可使个别酶活性增高,其代谢产物增多从而可引起疾病。例如有一类痛风(gout)是由于患者体内的磷酸核糖焦磷酸合成酶(phosphoribodsylpyrophosphate synthetase,PRPP-syn)结构变异,酶活性可升高3倍,致使磷酸核糖焦磷酸(PRPP)的生成大大加快(图4-24),结果由PRPP分解的终产物尿酸在体内大量增加,并从尿中排出,由于尿酸的溶解度低,过多的尿酸盐在结缔组织中沉积,出现痛风性关节炎,久后造成关节变形,大量尿酸在尿中沉积,可形成尿路结石。
目前已知PRPP-syn活性增高有三种情况:①酶分子合成正常,但每分子酶的活性增高2-3倍;②PRPP-syn与底物5’-磷酸核酮的亲和力增高;③酶与抑制物的亲和力降低。本病呈常染色体显性遗传。PRPP-syn酶基因定位于Xq22-q26.
酶活性过高导致产物过多引起的一类疾病称为生产过剩病(over-production disease)。
第三节 遗传多态现象
遗传多态现象(genetic polymorphism)是指在一个群体中,同时和经常存在的两种或两种以上的变异型或基因型,每种类型的频率比较高,一般认为每种变异型超过1%即可定为多态现象,不足1%的称为罕见变异型。
人类存在多种遗传多态现象(多态性),主要有染色体多态性、酶和蛋白质多态性、抗原多态性的DNA多态性五类。
1.染色体多态性染色体的多态性又称异态性(heteromorphism)是指正常人群中经常可见到各种染色体形态的微小变异。这种变异主要表现为同源染色体大小形态或着色等方面的变异。多态性是可遗传的,并且通常仅涉及一对同源染色体中的一个。例如表现的D和G组的随体增大、重复(双随体)或缺如,短臂的长短,1、9、16号染色体的次缢痕区加长或缩短,染色体着线粒区的荧光强度变异等。Y染色体长臂的长度变异,可大于F组,也可小于G组,这种变异可能有民族差异。
染色体多态性的临床意义尚不清楚,在产前诊断中,染色体多态性可分胎儿细胞和母体细胞;可探讨异常染色体不分离的来源,有利于对患者家庭进行婚育的指导。此外,可用于鉴定不同个体,对法医学中的亲权鉴定有一定的意义。
2.蛋白质多态性现在认为,人类结构蛋白质的多态性是一种普遍现象。例如结合珠蛋白(haptoglobin,Hp)是一种糖蛋白,其生理功能在于Hp和血红蛋白结合后不能透过肾小球膜,因而红细胞破坏后释出的血红蛋白不能被肾清除,既避免铁的大量丢失,也可保护肾免受损害。构成Hp分子的肽链有α和β链二种。Hp多态性主要是α链的遗传变异。α链有α1和α2二种。α1中又有αIs和αIF二种亚型,各由84个氨基酸组成,两者区别在于第54位氨基酸(αIF为赖氨酸,αIs为谷氨酸),因此可以推断这一区别是通过一次点突变形成。α2则由143个氨基酸组成。从α链一级结构分析,Hpα2是HpαIF的N端71个氨基酸和HpISC端的72个氨基酸连接而成,即发生了错误配对和不等交换,形成了与HbLepore相似的融合基因。Hpα1和α2肽链是由一对等位基因Hp1和Hp2所决定,基因定位于16q22.1,呈共显性遗传。因此有三种基本基因型:Hp1-1型为Hp=1/Hp1;Hp2-1型为Hp2/Hp1;Hp2-2型为Hp2/Hp2。除上述三型外。α还有其它变异型,由此组合成人群中Hp的多种多样的基因型和表现型。
常见的3种Hp类型在不同人种中分布不同,Hp1和Hp2的基因频率也各异,Hp2基因频率以亚洲人中最高(0.75),欧洲白人次之(0.60),非洲人最低(0.30~0.40)。
人血清运铁蛋白(transferrin,Tf)也是一种高度多态性系统。Tf基因位于3q21,已发现有30多种遗传类型(根据电泳迁移率快慢分型),也有明显的种族差异。
3.酶的多态性酶的遗传多态性表现为许多酶都在存在同工酶的现象。同工酶(isozyme或isoenzyme)是指分子结构不同的酶,可催化相同化学反应,这类酶称为同工酶。同工酶不仅可存在于不同个体。也可存在于不同的组织中,甚至在同一细胞的不同细胞器中有同工酶。
根据酶的多态性产生原因不同,大致可以分为三类:
(1)多座位同工酶(multiple loci determining isozyme):是指由不同基因座位决定的同工酶。例如乳酸氢酶(lactic dehydrogenase,LDH)有A、B两种亚基,分别由LDHA基因(定位于11p15-p14)和LKHB基因(定位一起12p12.2-p12.1)所决定。磷酸葡萄变位酶(phosphoglucomutase,PGM)的3个不同肽链分别由不同座位的基因编码。PGM1定位于1p22.1、PGM2定位于4p14-q12、PGM3定位于6q12。以LDH同工酶为例,LDH是A、B两种亚基不同比例组成的四聚体,依电泳速度可有5种表现型:LKH1(B4)、LDH2(B3A)、LDH3(B2A2)、LDH4(B1A3)及LDH5(A4)。如果A或B亚基再有不同突变,通过各种组合可以形成多种多样的变异类型。
(2)单座位复等位基因同工酶:是指同一座位上的不同等位基因所编码的酶蛋白。例如胎盘碱性磷酸酶(placental alkaline phosphatase,PLAP)是一种典型的复等位基因编码的酶,PLAP为二聚体,基因定位于2q37,依电泳速度可分3种变异型即慢(S)、中(I)、快(F)型,它们受3个复等位基因PLs、PLi及PLf控制,每种基因分别编码S、I、F三种同工酶(G6PD)和腺苷酸激酸(AK)的同工酶即属此类。
(3)翻译后同工酶:酶蛋白翻译产物经不同修饰反应也可产生不同分子形式的同工酶,称为翻译后同工酶(post-translational isozyme),又称次级同工酶(secondaryisozyme)。这些修饰反应包括基因的添加(磷酸化、乙酰化、甲基化等),酰胺键的水解,肽键的断裂和部分肽键的脱落,二硫键的形成和糖链的添加等。例如免肌醛缩酶(ALD)是由4个A亚基组成的纯聚体,但发育中,一部分肽链中羧基本末端第4位的天冬氨酸,从而A亚基形成α和β两种类型,故A亚基可为纯聚体也可为杂交体,但都有醛缩酶活性。
4.抗原多态性在人类遗传学应用较多的抗原有红细胞抗原系统和白细胞抗原系统。个体间抗原性差异是由基因突变产生。至今已发现20个红细胞血型系统,其中包括100多种红细胞抗原,比较重要的有:ABO、MNSs、Rh和Xg等血型系统。白细胞抗原系统中以人类的主要组织相容性复合体(mojor histocompatibility complex,MHC)中的人类白细胞抗原(humanleucocyte antigen,HLA)最引人注目,是目前所知人类最大的一个抗原多态系统。HLA的基因座位在6q21,已确认的HLA抗原特异性有148种,分属于A、B、C、D、DR、DP、DQ7个连锁基因座位。A座位具有24个抗原(即有24个复等位基因);B座位有52个;C座位有8个;D座位有26个;DR座位有20个;DQ座位有9个;DP座位有6个,因此,估计人群中可存在24×52×11×26×9×6=385,482,240种单倍型类型,即要在无亲缘关系人群中找到一条单倍型完全相同的人的机会只有约1/3.8×108。HLA多态性现象主要是由于产物不同而有不同的表现型。
5.DNA多态性在人群中不同个体之间的基因产物大多数是一致的,但每个个体在遗传上还是有所不同的,这种个体之间的差异从本质上讲是DNA碱基顺序存在的差异,它是通过用内切酶切割不同个体的基因组DNA出现不同长度的片段而被发现的,并通过孟德尔方式遗传,称为DNA多态性。DNA多态性的来源,它在医学中的应用价值将在第十三章中阐述。
第五章 多基因病
第一节 多基因遗传与数量遗传
多基因遗传(polygenicinheritance)是指生物和人类的许多表型性状由不同座位的较多基因协同决定,而非单一基因的作用,因而呈现数量变化的特征,故又称为数量性状遗传。多基因遗传时,每对基因的性状效应是微小的,故称微效基因(minor gene),但不同微效基因又称为累加基因(additive gene)。多基因遗传性状除受微效累加基因作用外,还受环境因素的影响,因而是两因素结合形成的一种性状,因此,这种遗传方式又称多因子遗传(multifactorical inheritance)
多基因遗传的数量性状(quantitativecharacter)为连续变异的性状,可以正态分布曲线表示。人的身高、血压和智力都是多基因性状,而单基因的质量性状(qujalitatie character)则呈不连续变异。图5-1表示单基因的侏儒症身高的变异分布。该病患者的平均身高仅130cm。这类人的家系成员身高决定于基因型AA、Aa和aa,变异的分布呈不连续的特点,即变异个体可明显区分为几个群(即曲线的几个峰),这各人的身高表现为一种质量性状,群体间差异显著。而正常人的身高是基因性状,身高平均为165cm,变异在群体中是连续的,曲线只有一个峰即平均值。人身高由矮到高是逐渐过渡,很矮和很高的两种极端的人只是极少数,大多数人身高接近平均值,这种变异的曲线呈正态分布(图5-2)。
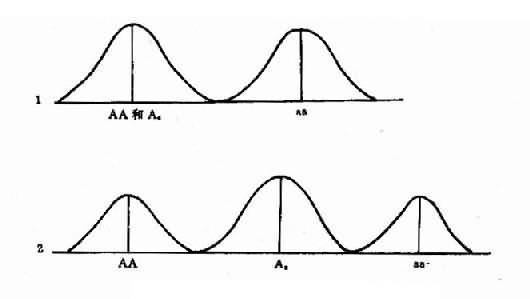
图5-1 侏儒症身高的变异分布图
1.完全显性;2。不完全显性
图5-2 正常人群身高的变异分布图
由此可见,侏儒症患者的身高是一种单基因遗传病的表型性状,而正常人身高则是既受多基因遗传控制,又受环境因素影响。对大多数人来讲,控制身高的基因是微效的和累加的。而极少数遗传病(如Klinefelter综合征、生长激素缺乏症等)对身高的影响是小的,在整个群体的分布曲线中几乎没有作用。分析身高遗传表明子代平均身高更加接近群体的身高平均值,而不是双亲的身高的平均值。统计学应用于遗传学中时,数量性状遗传在子代中出现少量极端表型个体是正常的。因此,从遗传基础来看,一级亲属平均有50%相同的基因,二级亲属平均有1/4的基因相同,三级亲属有1/8的基因相同,如表5-1所示。
表5-1 不同亲属关系的基因比例
| 亲属关系 | 基因比例 |
| 单卵双生 | 1 |
| 一级亲属(异卵双生,双亲,同胞,子女) | 1/2 |
| 二级亲属(祖父母,叔,伯,舅父,侄子女、孙子女) | 1/4 |
| 三级亲属(表堂兄妹、曾祖父母、曾孙子女) | 1/8 |
多基因遗传具有3个特点:①两个极端变异(纯种)个体杂交后,子1代大部分为中间型,具有一定变异范围,是环境影响。②两个中间型子1代杂交后,子2代大部分为中间型,但其变异范围要比子1代广泛,也可出现极端的个体。这除环境因素外,基因的分离组合也有作用。③在随机杂交的群体中,变异范围很广,然而大多数个体接近中间型极端个体很少,环境与遗传因素都起作用。
第二节 多基因病
一些常见的先天畸形(congenitalmaiformation)和常见而病因复杂的疾病,其发病率一般都超过1/1000,疾病的发生都有一定的遗传基础,并常出现家族倾向,但不是单基因遗传,患者同胞的发病率不遵循1/2或1/4的规律,大约仅1%-10%,表明这些疾病有多基因遗传基础,故称为多基因病(polygenic disease)。正因为它们的遗传基础很复杂,属于复杂遗传病(complexgenetic disease)。
1.易患性与发病阈值多因子遗传性状和多基因病是由基因与环境因素共同作用的。决定一个个体是否易于患病的基础,称为易患性(liability)。易患性的变异与多基因遗传性状一样,在群体中呈正态分布,即群体中大多数个体的易患性近似平均值,易患性很高或很低的都很少。当一个体的易患性达到一定程度,即阈值(threshold),这种个体就要患病。因此,易患性的变异在群体中的分布就被阈值分为两部分:大部分为正常个体,小部分为患者。阈值代表在一定条件下患病所必需的、最低的易患基因的数量(图5-3)。
图5-3 群体中易患性变异与阈值图解
虽然就一个个体来说,易患性难以测定,只能依其婚后所生子女病情况作出粗略估计,但一个群体的易患性则可由该群体的发病率(即超过阈值部分)作出估计。其估量的尺度以正态分布的标准差为单位,就是在正态分布中,以平均值为0,在±1个标准差(0)范围内的面积占曲线内总面积的68%,以外的面积的占32%,两边各占16%。如此类推,在±2个标准差以内面积占总面积的95.4%,标准差以外的面积占4.6%,两边各占2.3%;在±3个标准差时,标准差以内面积占总面积的99.74%,以外面积占0.26%,两边各占0.13%。多基因病的易患性阈值与平均值距离越近,其群体易患性的平均值越高,阈值越低,则群体发病率也越高。反之,两者距离越远,其群体易患性平均值越低;阈值越高,则群体发病率越低。因此,可从群体发病率的高低计算出阈值与平均值之间的距离。图5-4表示易患性阈值和平均值距离与发病率的关系。
2,遗传率遗传率是为了衡量多基因遗传中遗传因素与环境因素两者的相对作用大小而提出的。遗传率(或遗传度,heritability)是指在疾病发生中,遗传基础所起作用的大小。遗传率一般用百分率(%)来表示。一种遗传病如果完全由遗传基础决定,其遗传率就是100%,当然这种情况很少见。在多基因病中,遗传率可高达70%-80%,这表明其遗传基础在决定易患性变异和发病上起着重要作用,而环境因素的影响较小;反之,遗传因素是次要的。遗传率的计算可用变量分析方法进行。
图5-4 易患性阈值、平均值距离与发病率的关系图解
3.某些先天畸形大约2%的新生儿存在严重的畸形,但多数尚能存活,其中很多畸形,如泌尿生殖系、脊椎和心脏的缺陷,在童年或青少年时已可鉴别诊断。
先天畸形的病因既有遗传因素,也有环境因素,还有两者共同的作用,故后者属于多基因遗传。后者是这些畸形最常见的病因,但在所有畸形中至少有40%的确实原因尚不知晓。少数遗传性畸形已证明是由于生化异常。如Ⅰ型成骨不全,已从患者皮肤成纤维细胞中测知其Ⅰ型胶原中的异常的a2链,这是骨骼发育异常的常染色体显性遗传病。还有诸多染色体异常引起的先天畸形不再赘述。
多基因遗传引起的畸形,遗传和环境因素都有影响,诸如社会经济原因对母体及其子宫的影响均可导致畸形的产生。常见的这类先天畸形有脊椎裂、唇裂、腭裂、短指或缺指、先天性心脏病、先天性幽门狭窄、先天性髋脱臼、先天性肾缺乏、先天性巨结肠症等。举几例简述如下。
(1)先天性心脏缺陷(congenital heart defects, CHDs):是一种很常见的先天畸形,在新生婴儿中发病率4%-8%,其原因是异质性的,有单基因或染色体异常引起的,也有因风疹病毒感染母体或糖尿病而引起的,大多数的病因不清。由于其类型较多,群体发病率及经验风险率也不一样。据多因子阈值模型所示,其一级亲属风险率是多因子性状的群体发病率的平方根据。表5-2示各种心脏缺陷畸形在群体和同胞中的发病率(表5-2)。
表5-2 主要先天性心脏缺陷在群体及同胞中的发病率
| 心脏缺陷 | 群体发病率 | 同胞中频率(%) | 群体发病率的平方根 |
| 室间隔缺损 | 1/575 | 4.3 | 4.2 |
| 动脉导管未闭 | 1/1200 | 3.2 | 2.9 |
| 房间隔缺损 | 1/1500 | 3.2 | 2.6 |
| 主动脉狭窄 | 1/2250 | 2.6 | 2.1 |
(2) 先天性单侧缺如(congenital absence of one kidney):约1/700婴儿患此病。大多数患者无症状,但伴发输尿管和结肠畸形者发病率高。如输尿管肾盂连接部狭窄,易感染,或发生高血压等。受累女性可伴有双角子宫或同侧半子宫缺如,受累男性伴有同侧输精管缺如。患者父母所生的子女侧肾发育不全的风险增高。
(3)短指畸形E型(brachydactyly,type E):由于骨骺未成熟闭合,病人手和脚变短并伴有第1、4和5掌骨、骨和拇指及趾的趾骨变短。患者身材矮小,青少年还伴有严重高血压。
(4)脊柱裂(spina bifida):受累的男性女性都可活到成年,通常智力正常,但都可能不育。受累成人的子女具有神经管缺陷的风险大约增高3%。
(5)先天性巨结肠(Hirschsprung’s disease):一般男性多于女性,但女性病人的子女中受累者风险较高。最近,已于染色体10q上将此病有关基因克隆。
由多基因所致的畸形患者,同胞及子女的患病风险增高,再发风险一般在1%-10%之间,比一般群体中畸形发病率高10-40倍,见表5-3。
表5-3 多基因遗传畸形患者的子女受累的风险
| 畸形 | 子女受累风险(%) | 一般群体发病率(%) |
| 先天巨结肠 | 2.0 | 0.02 |
| 尿道下裂 | 6.0 | 0.8 |
| 马蹄内翻足 | 1.4 | 0.13 |
| 先天性髋关节脱位 | 4.3 | 0.8 |
| 室间隔缺损 | 4.0 | 0.2 |
| 先天性幽门狭窄 | 4(受累父亲) | |
| 13(受累母亲) | 0.3 | |
| 腭裂 | 6.2 | 0.3 |
| 脊柱裂 | 2.0 | 0.14 |
4.某些多基因遗传的常见病成人许多常见的慢性疾病,如原发性高血压、糖尿病、动脉粥样硬化、冠心病、精神分裂症、哮喘病等都属于多基因病。这类疾病发病既有多基因遗传基础又有环境因素。如在任何一个体中,有特殊的大量致病基因,潜在发病危险性就大。当一个个体继承了危险基因的正常组合,他(她)们通过“危险阈值”时,环境因素就可决定疾病的表达和严重性,如图5-5。
要使其家庭成员患同一种疾病,该个体必须继承相同或极相似的基因组合,这种情况发生的可能性在一级亲属中明显大于较远的亲属。多基因遗传的易患性必须服从于基因控制特定功能的特异蛋白质合成这个基本事实。
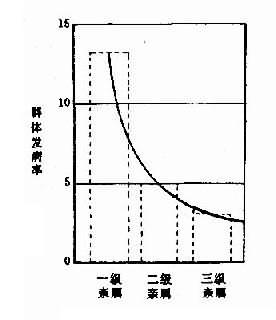
图5-5 多基因病在群体及各级亲属中的闰病率
(1)冠状动脉病(coronary artery disease, CAD):大多数病例为有明显环境因素作用的多基因遗传病。CAD不仅有单基因病因,还伴有许多其它非遗传性和遗传风险因素。遗传因素如男性、家族史、高脂血症、高血压、糖尿病、肥胖症等;非遗传因素如吸烟、不活动、精神紧张等,因而增高了患该病的风险。
CAD患者大多在45岁以上,男性有较高的风险,这在群体和受累家族中都是如此。而当女性为先证者时,其亲属中的再发风险较高(表5-4)。
表5-4式CAD男女性先证者与死亡风险关系
| 亲属关系 | 死亡风险 |
| 男性先证者的男性亲属 | 1/12 |
| 男性先证者的男性亲属 | 1/36 |
| 女性先证者的女性亲属 | 1/10 |
| 女性先证者的女性亲属 | 1/12 |
(2)高血压病(hypertension):它是一种复杂的多基因遗传病。在我国高血压病的患者高达8000余万人,近十年来升高约25%。高血压病具有家族聚集现象和复杂的遗传方式,其遗传率约为30%-60%。分子遗传学研究表明,该病和两个重要的基因,即血管紧张素转化酶基因(angiotensin converting enzyme,ACE1)和血管紧张素原基因(angiotensinogen,AGT)有关。ACE1的产物是血管紧张素Ⅱ(angiotensinⅡ),它与血管的构建和细胞生长有关,并决定血压、体液和离子的稳定性。血管紧张素原是血管紧张素Ⅱ的前体,AGT的增加,也伴随血管紧张素Ⅱ相应提高。某些激素对血压的影响也是通过AGT而实现的。最近证明AGT基因与高血压连锁,高血压时局部AGT水平增高。在高血压病的发病中,环境因素(精神紧张、高盐食物等)也是众所周知的。
(3)糖尿病(diabetes mellitus):本病是胰岛缺乏引起的常见代谢性疾病。按对胰岛素的需要量分为胰岛素依赖型糖尿病(IDDM)和非胰岛素依赖性糖尿病(NIDDM)两个类型。其特征为血糖升高、糖尿、糖耐量降低和胰岛素释放反应异常。临床上患者多食、多饮、随病程进展可出现心血管、肾、眼及周围神经等病变。一般认为是多基因病。近年用糖尿病大白鼠模型研究,认为该病可能与3个以上基因有关。
目前对多基因的常见复杂病的遗传基础研究非常引人注目,且进展迅速。许多事实证明多因子疾病中存在着单基因的遗传形式(monogenic forms of multifactorialdiseases)这是多基因遗传病研究非常重要的发展趋向。
5.多基因病的一些特点综上所述,可见多基因病有如下特点:
(1)家族聚集倾向,但无明显的遗传方式。因为在系谱分析中,它们不符合单基因遗传方式,同胞中发病率远低于1/2或1/4,既不符合常染色体显性和隐性遗传,也不符合X连锁遗传,但这些疾病及其在子代的再发风险,确实表现出家族性聚集倾向。
(2)随亲属级别的降低,患者亲属发病风险迅速下降,在发病率低的疾病,这个特点更为明显。表5-5和图5-6说明一些多基因病患者不同级别亲属发病风险的比较和由阈值模型得出亲属级别发病风险的理论曲线。
表5-5 某些多基因遗传病患者不同级别亲属的发病风险比较
| 疾病 | 群体发病率 | 发病风险 | |||
| 一卵双生 | 一级亲属 | 二级亲属 | 三级亲属 | ||
| 唇裂士腭裂 | 0.001 | ×400 | ×40 | ×7 | ×3 |
| 足内翻 | 0.001 | ×300 | ×25 | ×5 | ×2 |
| 神经管缺损 | 0.002 | ×8 | ×2 | ||
| 先天性髋关节脱臼 | 0.002 | ×200 | ×25 | ×3 | ×2 |
| 先天性幽门狭窄 | 0.005 | ×80 | ×10 | ×5 | ×1.5 |
(3)近亲婚配时,子女患病风险增高,但不如常染色体隐性遗传显著,这可能与多因子的积累效应有关。
(4)畸形越重,再现风险越大,这说明遗传因素起着重要作用。
(5)当一种多基因性状频率在不同性别有明显差异时,表明发病率高的性别其阈值低,发病率低者其阈值高。图5-7表明先天性幽门狭窄由于性别不同的阈值差异的易患性分布,
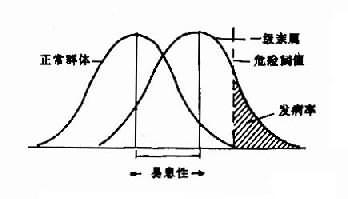
←易患性→
图5-6多基因遗传阈值模型
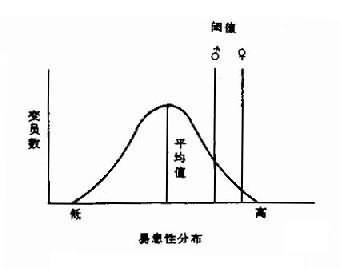
易患性分布
图5-7 有性别差异阈值的易患
即群体发病率低的性别患者,只有他们带有相当多的易患性基因时,其易患性才能超过阈值而发病。如果他们已发病,就表明他们已带有相当多的易患性基因,其后代中发病风险将较低,尤其是与其性别相反的后代。群体发病率高的性别患者,其后代中发病风险将较低,尤其是发病率低的性别的后代。先天性幽门狭窄患者,男性发病率是女性的5倍(男0.005,女0.001)。如为男性患者,儿子发病风险为5.5%,女儿发病风险为2.4%;相反,如为女性患者,她儿子的发病风险为19.4%,女儿风险为7.3%。
第一节 多基因病复发风险估计
在相当多的基因病中,其群体发病率为(0.1%-1%),遗传率为70%-80%。患者的一级亲属的发病率(f)近似于群体发病的平方根,即f=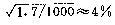 ≈4%。如果群体发病率过高或过低,则上述Edward公式不适用。要了解一群体发病率、遗传率和患者一级亲属发病率的关系,则需要看图5-8。如原发性高血压的群体发病率约为6%,遗传率为62%,患者一级亲属的发病风险从图5-8即可查出约为16%。
≈4%。如果群体发病率过高或过低,则上述Edward公式不适用。要了解一群体发病率、遗传率和患者一级亲属发病率的关系,则需要看图5-8。如原发性高血压的群体发病率约为6%,遗传率为62%,患者一级亲属的发病风险从图5-8即可查出约为16%。
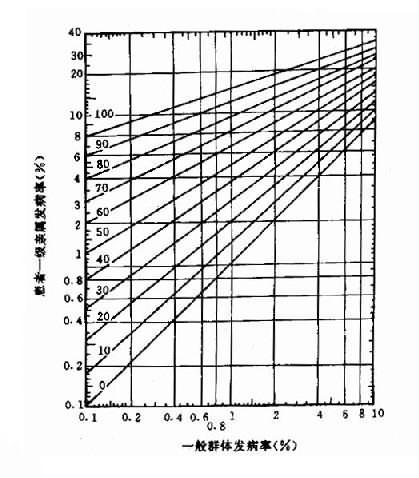
图5-8 一般群体发病率、遗传率与患者一级亲属发病关系图解
在估计多基因病发病风险时应注意几个问题:
1.基因的积累效应与再发风险一般而论,一个家庭中患病人数越多时,意味着再发风险越高。例如一对夫妇已有一个唇裂患儿,再次生育的再发风险为4%,若又生出一个这样患者,则表明夫妇二人都带有较多的易患基因,虽然他们本人未发病,但其易患性极为接近阈值,这就是基因效应所致,再次生育的再发风险将增加2-3倍,即近于10%。表5-6为Sith研制的一个表格,通过双亲是否为患者及其同胞中已发生该病患者人数来估计再发风险。
表5-6 多基因病再发风险估计
| 双亲患病数 | 1 | 2 | ||
| 一般群体发病率(%) | 遗传率(%) | 患者同胞数 | 患者同胞数 | 患者同胞数 |
| 0 1 2 | 0 1 2 | 0 1 2 | ||
| 1.0 | 100 80 50 | 1 7 14 1 6 14 1 4 8 | 11 24 34 8 18 28 4 9 15 | 63 65 67 41 47 52 15 21 26 |
| 0.1 | 100 80 50 | 0.1 4 11 0.1 3 10 0.1 1 3 | 5 16 26 4 14 23 1 3 9 | 62 63 64 60 61 62 7 11 15 |
2.病情严重程度与再发风险多基因病中基因的累加效应还表现在病情的程度上。因为病情严重的患者必定带有更多的易患基因,其父母也会带有较多的易患基因使易患性更接近阈值。所以,再次生育时的再发风险也相应地增高。例如,只有一侧唇裂的患者,其同胞的再发风险为2.46%,若一侧唇裂合并腭裂的患者,其同胞的再发风险为4.21%,而两侧唇裂合并腭裂的患者,其同胞的再发风险则高达5.74%。
3.性别与再发风险这在多基因病特点中已讲过,不再重复。
因此,在估计多基因病再发风险时,必须全面考虑上述各种情况,进行综合判断,才能得出符合实际的数据,有效地指导实践。表5-7是一些常见多基因病的遗传率及再发风险的举例说明。
表5-7 某些常见多基因病的遗传率
| 病名 | 群体发病率(%) | 患者一级亲属发病率(%) | 男:女 | 遗传率(%) |
| 唇裂±腭裂 | 0.17 | 4 | 1.6 | 76 |
| 腭裂 | 0.04 | 2 | 0.7 | 76 |
| 先天性髋关节脱位 | 0.1-0.2 | 4 | 0.2 | 70 |
| 先天性幽门狭窄 | 0.3 | 男性先证者2 | 5.0 | 75 |
| 女性先证者10 | ||||
| 先天性畸形足 | 0.1 | 3 | 2.0 | 68 |
| 先天性巨结肠 | 0.02 | 男性先证者2 | 4.0 | 80 |
| 女性先证者8 | ||||
| 脊柱裂 | 0.3 | 4 | 0.8 | 60 |
| 无脑儿 | 0.5 | 4 | 0.5 | 60 |
| 先天性心脏病(各型) | 0.5 | 2.8 | - | 35 |
| 精神分裂症 | 0.1-0.5 | 4-8 | 1 | 80 |
| 糖尿病(青少年型) | 0.2 | 2-5 | 1 | 75 |
| 原发性高血压 | 4-8 | 15-30 | 1 | 62 |
| 冠心病 | 2.5 | 7 | 1.5 | 65 |
| 支气管哮喘 | 4 | 20 | 0.8 | 80 |
| 胃溃疡 | 4 | 8 | 1 | 37 |
| 强直性脊柱炎 | 0.2 | 男性先证者7女性先证者2 | 0.2 | 70 |
第六章 群体遗传学
群体(popualtion)是指同一物种生活于某一地区并能相互杂交的个体群。人种大约50多亿的成员分为许多不同的亚群体,其中最大的通常称为种族(races)。不同的群体,作为大群体其基因库也是彼此不同的。人类有三个主要人种区分:高加索人.黑人各亚细亚人,但他们又都有许多遗传上不同的亚群,种与亚群体存在主要差异的遗传基础是突变。
群体遗传学(population genetics)是研究群体的遗传变化规律的学科,研究群体中基因的分布.基因频率和基因型频率的维持和变化的科学。医学研究群体遗传是要探讨遗传病的发病频率.遗传方式及其基因频率和变化的规律,从而了解遗传病在群体中的发生和散布的规律,为预防、监测和治疗遗传病提供重要的信息和措施。因此有人又称之为遗传流行病学(genetic epidemiology)。
第一节 群体中的遗传平衡
群体中的遗传基因和基因型需要保持平衡,才能保证人种的世代繁殖。而一个群体所具有的全部遗传信息,亦即含有在特定位点的全部等位基因称为基因库(gene pool)。个体的基因型只代表基因库的一小部分。在研究群体变化、群体中遗传病的变化时,需要了解遗传的发病率及其遗传性状,这就要了解某一基因的存在,分析并计算基因频率。
一、基因频率
基因频率(genefrequency)是指群体中某一基因的数量。基因频率也就是等位基因的频率。群体中某一特定基因的频率可以从基因频率(genotype frequency)来推算。如人们熟悉的人的MN血型,它是由一对共显性等位基因M和N所决定,产生3种基因型M/M、M/N和N/N,而相应的表型是M、MN和N,而且比例是1/4M、1/2MN和1/2N。这个原理可以推广到一般群体内婚配,如以群体中MN表型(基因型)的具体样本数被所观察到总数相除即可得到(转换)相对频率数。表6-1示某一群体的MN血型的相对基因频率,再用此观察到的相对来估算M和N在群体中等位基因的频率。该群体中总人数为1419人,其中M血型(基因型M/M)392人,N血型(基因型N/N)320人,MN血型(基因型M/N)707人,按此可直接推算出M的等位基因频率为:
2×392——707/1419×2=0.53
表6-1 某群体的MN血型的相对基因频率
| 表型 | 基因型 | 个体数 | 相对频率 |
| M | M/M | 392 | 0.276 |
| N | N/N | 320 | 0.226 |
| MN | M/N | 707 | 0.498 |
| 总计 | 1419 | 1.000 |
同样,N的基因频率为1-0.53=0.47。因为两个等位基因的相对频率相加必须等于1。换言之,设M的基因频率为p,N的基因频率为q,p+q=1(或100%)。所以,在研究一个等位基因在群体中的频率时,就要考虑到基因库。基因库实际上就是一个群体的全部等位基因。如果知道MN两个等位基因在基因库中的相对频率是0.53(M)和0.47(N),而群体中样本人数是1419,那么这3种组合的等位基因(基因型)的相对比例就得以知道。
即2M=0.53×0.53=0.281
2N=0.47×0.47=0.221
MN=(0.53×0.47)+(0.47×0.53)=0.498
上述例子以一般公式表示:p、q分别为两个等位基因频率,等位基因的3种组合的频率应为:
二、遗传平衡定律
表型、基因型和等位基因频率之间关系的一个重要原则就是基因型的比例在世代传递中不会改变。因此,群体中个体的等位基因频率的分布比例和基因型的分布比例(频率)世代维持恒定。这是群体遗传学的一个基本原则,是Hardy和Weinberg于1908年应用数学方法探讨群体中基因频率变化所得出的结论,即遗传平衡定律(又称Hardy Weinberg定律)。意即一个群体符合这种状况,即达到了遗传平衡,也就是一对等位基因的3种基因型的比例分布符合公式:p2+2pq+q2=1,p+q=1,(p+q)2=1.基因型MM的频率为p2,NN的频率为q2,MN的频率为2pq。MN:MN:NN=P2:2pq:q2。MN这对基因在群体中达此状态,就是达到了遗传平衡。如果没有达到这个状态,就是一个遗传不平衡的群体。但随着群体中的随机交配,将会保持这个基因频率和基因型分布比例,而较易达到遗传平衡状态。
1.遗传平衡的条件要维持群体的遗传平衡需要一定的条件,或者说这种遗传平衡受一些因素的影响。这些条件和因素是:①群体要很大,不会由于任何基因型传递而产生频率的随意或太大的波动;②必须是随机交配而不带选择交配;③没有自然选择,所有的基因型(在一个座位上)都同等存在,并有恒定的突变率,即由新突变来替代因死亡而丢失的突变等位基因;④不会因迁移而产生群体结构的变化。如果缺乏这些条件则不能保持群体的遗传平衡。
2.群体的遗传平衡和等位基因频率推算由于人类群体中大多数遗传性状都处于遗传平衡状态,据此可计算等位基因频率。如已知某种常染色体隐性遗传病(白化病)在一特定人群中频率,就能计算这个异常基因的携带者和基因频率。白化病发病(aa)(q2)的频率为1/10000,即其基因型频率,则致病基因(a)频率=[]=1/100=0.01;基因A的频率=1-0.01=0.99;而杂合子携带者的频率为2pq=2×99/100×1/100≈1/50。因此,在比例中,每个受累的个体将有200个左右在临床上无症状的携带者。
常染色体显性遗传病,如并指症,在一个群体多为杂合子(Aa)发病。杂合子(H)的频率为2pq,由于q值大,近于1,故H=2p,p=1/2H。因此,只要知道杂合子发病率,就易求得基因A的频率。如并指症的发病率为1/1000,H=1/2000,p=1/2H=1/4000,即致病基因A的频率为0.000025。
X连锁遗传性状也符合遗传平衡定律。因为女性中的基因频率及基因型频率分布和常染色体相同,而男性是半合子,其基因频率.基因型频率与表型频率相同,如表6-2所示。
表6-2 X连锁红绿色盲的基因型及基因频率
| 性别 | 基因型 | 表现型 | 基因型频率 | 发病率 |
| 男 | XAY XaY | 正常 色盲 | p q | 0.92 0.08 |
| 女 | XAYA XAXa XaXa | 正常 正常 色盲 | p2(纯合) 2pq(杂合) q2 | (0.922)=0.846 2(0.92)(0.08)=0.147 (0.08)2=0.0064 |
至于罕见的常染色体隐性遗传病纯合子患者频率(q2)很低,所以2pq=2(1-q)q=2q-1q2=2q,即杂合子携带者频率(2pq)约为致病基因频率的2倍,故致病基因多以携带者的方式存在于一个群体中。在前述白化病中,发病率1/10000,q为0.01,携带者频率约为2q,即2×0.01=0.02(1/50)。
第二节 突变和选择
1.突变率及其表达方式突变有其分子基础。因为自然界中普遍存在着突变,每个基因都有一定的突变率(mutation rate)。一般用每代中每一百万个基因中发生的突变数来表示,即n×10-6/基因/代。突变对遗传平衡的影响有正负两个.如一对等位基因A(显性)和a(隐性),由显性基因(A)突变为隐性基因(a)的过程。称为正向突变(forward mutation),其突变率以u表示;由隐性基因(a)突变为显性基因(A)的过程,称为回复突变(reverse mutation),其突变率以v表示。设A基因的频率为p,a基因的频率为q,A基因的突变率为u,a基因的突变率为v,这样,每一代中就有pu或(1-q)u的基因A突变为a,也有qv的基因a回复突变为A。若(1-q)u>qv,则基因a的频率将增加,反之,(1-q)u
(1-q) u=qv
u=qu=qv
u=qu+qv
u=q(u+v)
q=μ/u+v
p=v/u+v
在此情况下,突变频率完全由u和v 来决定。突变有时导致一个基因功能的丧失或变化。大多数可检测的突变是有害的,因此有突变就会面临选择,如果选择是中性,即中性突变(neutral mutaiton)。这种突变型既无害处亦无益处,选择性不显著。如我国汉族人群中,对苯硫脲(PTC)缺乏尝味能力的味盲(tt)的频率为9%,味盲基因(t)的频率为0.03,这里 =0.03。此PTC味盲基因(t)就可看成是来源于中性突变。它对人体既无特殊益处,也无明显的害处,选择作用也不明显。
=0.03。此PTC味盲基因(t)就可看成是来源于中性突变。它对人体既无特殊益处,也无明显的害处,选择作用也不明显。
2.选择和适合度选择(selection)是指自然选择。选择对遗传平衡的作用是增加或减少个体的适合度(f)。适合度(fitness)是指个体在一定条件环境下,能生存并传递其基因于下代的能力。可以说是生存和生育率联合效应的最后结果,可用相同环境中,不同个体的相对生育率(fertility)来衡量。例如丹麦对软骨发育不全患者的调查,该病是常染色体显性遗传病,有矮小、指短、头大、低鼻梁、前额大等体征。在108例患者中,共生育了27个儿女,而他们的正常同胞475人中共生育了582个子女,如以正常人的生育率为1,该病患者的相对生育率(f)=(27/108)/(528/457)=0.2。这样表明在软骨发育不全患者中的适合度降低了,同时也说明相对生育率即代表适合度。人们可用类似方法求得其它遗传病患者的适合度。
选择系数(压力)(selectioncoefficient or pressure)是与适合度有关的概念,并以此来表示选择的作用,一般用s表示,它表明在选择作用下,降低的适合度。s与f的关系是:s=1-f。如软骨发育不全患者的适合度为0.02,其选择系数s=1-0.2=0.8。
3.选择对隐性基因、显性基因和X连锁基因的作用及有关突变率的计算 在一定条件下,一个群体的突变率可明显增高,形成突变压力(mutation pressure),使某个基因频率增高。另一方面,受某种环境条件的影响,某些突变型选择作用,使突变基因的频率降低,即选择压力。因此,一个群体保持遗传平衡,就是突变压力和选择压力之间达到平衡,这样,使群体的遗传结构趋于稳定,达到平衡状态。
(1)选择对显性基因的作用:在一个遗传平衡群体中,选择对显性基因作用较为明显,因为有显性基因的个体(AA和Aa)都可受到选择的作用。当选择对显性基因A不利时,杂合子Aa和纯合子AA都会被淘汰。这样,基因A最终会从群体中消失。这时如要达到遗传平衡,就要靠基因a突变为基因A来补偿。这样显性基因突变率可按下列公式求出: v为突变率,s为选择系数,p为基因A的频率,患者多为杂合子(H),其频率为2pq,由于p值很小,q≈1,2pq=2p。
p=1/2H,H=2p,故v=s×1/2H
也就是说每一代中基因频率因选择而降低为s× 1/2H,而在一个遗传平衡群体中被淘汰的部分将由突变率(v)来补偿,即按v=s×1/2H公式求出显性基因的突变率。例如软骨发育不全侏儒症,在丹麦哥本哈根市医院出生的94 075名儿童中,患该病的有10人,其发病率为10/94,075=0.0001063。又已知该病适合度为0.02,则其选择系数s为0.08。代入公式v=s×1/2H=0.08×0.5×0.0001063=42.5×10-6/代。此外,还有证据表明,随父亲年龄的增长,某些常染色体显性遗传的基因突变率增高。
(2)选择对隐性基因的作用:如只有隐性基因(a)纯合(aa)时才发病而面临选择。由于隐性基因大都可以杂合状态在群体中维持很多世代,因此,选择的作用对隐性基因频率的降低很慢,如选择对其不利时,最终将从人群中消失。而隐性基因频率在人群中基本恒定,这可能是基因A突变为基因a,以补偿因选择而被淘汰的有害隐性基因aa,即在一个遗传平衡群体中,被淘汰的部分将由突变率(u)来补偿。选择系数为s,每一代在基因频率的减少将为sq2,故u=sq+2,按此公式可以隐性基因的突变率。例如苯酮尿症(PKU)在我国人群中的发病率为1/16,500(0.000060),其适合度f为0.15,s=0.85,故u=sq2=0.85×0.00006=0.000051=51×10-6/代。可见此突变率相当高,因为PKU的适合度过低。
(3)选择对X连锁基因的作用;由于男性是半合子,X连锁隐性基因只要是致病基因便可表达而面临选择,所以致病基因频率q与就是男性发病率。而女性携带者的频率近于致病基因频率的2倍,但携带者的表型正常,不受选择影响。因此,从整个群体来说,男性致病基因频率只占全部致病基因量的1/3,选择系数为s,每代中因选择而被淘汰的致病基因为1/3sq,而被淘汰的部分由突变率(u)来补偿,故u=1/3sq。
例如甲型血友病男性发病率为0.00008,适合度f为0.25,则s=0.75,代入上式:u=1/3×0.75×0.00008=0.00002=20×10-6/代,这样的突变率也是较高的。假肥大型肌营养不良(DMD)的突变率更高,u=20-100×10-6/代。
4.选择压力的变化对遗传平衡的影响既然维持群体遗传平衡受选择和突变及其压力之间平衡的制约,则选择压力的变化必然要影响群体的遗传结构。选择压力的变化主要有两方面:一是选择压力的增强,二是选择压力的降低,这两种情况可影响群体中某一表型的适合度增高或降低,从而使表型相应的等位基因的频率也发生改变。
(1)选择压力的增强:即选择压力越大,选择系数越高,引起基因频率变化越快。对常染色体显性遗传病,杂合子患者(Aa)的适合度为0,s=1,则群体中致病基因A的频率p在一代中间即可降低为0,下一代中致病基因频率将靠突变率来维持,因上一代的致病基因已被选择而淘汰,因此,基因频率的变化较迅速。对常染色体隐性遗传病,选择压力即使增强,纯合隐性患者aa的频率为q,基因AA的频率为p2,Aa的频率为2pq,aa的频率为q2。在aa的适合度为0,s=1时,则群体中基因a,只存在于杂合子Aa中。计算隐性致病基因频率降低所需世代数可按下列公式进行。
q1=q/1+q q2=q1/1+q2以后的世代为qn=q/1+qn
n=1/qn-1/q(n表示世代)
例如,常染色体遗传病白化病,其致病基因a在群体中的频率为0.01,在选择压力增强时,使该病患者(aa)不能生育,需经过多少世代才能使其基因频率降低为0.005呢?q=0.01=1/100,代入公式:n=1qn-1/q=1/(1/200)-1/(1/100),n=200-100=100代。如果每世代以25年计算,则要经过2500年才能使基因频率降低一半,因此,选择压力的改变对隐性基因频率的变化是很缓慢的。
(2)选择压力放松(relaxation fo selection pressure):由于选择压力的降低使致病基因频率增高,而导致遗传发病率增高。随着医学发展和诊治技术的进步,将有许多遗传病可以治疗或延长患者生命,他们可活到生育年龄,并结婚生育,这样是否会增加群体中遗传病的发病率呢?
对显性遗传病来说,若是致死的,其发病率完全由突变来维持。若患者被治愈就和正常人一样结婚生育,这样,经过一代后发病率将增加1倍,而后代各代中将以同样数量增加。当然,该病病例也将逐渐增加,人们从遗传型视网膜母细胞瘤就已看到这种变化趋势。
对隐性遗传病来说,若是致死的,其发病率在完全放松选择的情况下,缓慢上升。例如,苯酮尿症的发病率约为1/10000,基因频率为0.01,突变率为50×10-6/代,该病可用低苯丙氨酸食品治疗。假定被治愈的患者生育率与正常人相同,即在完全放松选择后,致病基因频率要经达200代才能增加1倍,即0.01+0.5/100000×200=0.02。这时的发病率比原来增高4倍,这种变化要经200代即5000年后才能达到,所以上升是很缓慢的。
对X连锁遗传病来说,只有男性患者才面临选择。若该病是致死的,在完全放松选择后,也要经过3代,致病基因频率才可增加1倍,即男性患者的发病率增高1倍。
总之,在选择放松情况下,遗传的发病率会有所上升,但其速度是缓慢的。现今分子生物学的迅速进展,先进医疗技术不断涌现,其中基因治疗展示了美好的前景,人们有理由不必选择压力的放松增高遗传病的发病率而忧虑。
5.选择与群体中的平衡多态性当选择压力向两个方面进行时,一方面是有害等位基因的维持,另一方面是它们的消除。也就是在一个群体中,只要等位基因存在,就会有两种或两种以上的基因型,其中最低的基因频率也不能仅用突变来维持,各基因型达到了遗传平衡,这种情况称为平衡多态性(balanced polymorphism)。
人类许多基因座位都存在着多态性,最稳定的多态性是突变基因由于有选择优势而形成。例如镰形细胞贫血症,在非洲黑人中,其纯合患者(HbsHbs)可高达4%,HbstHbA的基因频率分别为0.2和0.8。纯合子一般死亡。杂合子在群体中可高达32%,这是因为杂合子的血蛋白结构的有抗疟性,其适合度略高于正常人(HbAHbA)。这样,在恶性疟疾流行时,杂合子具有选择优势。杂合子的这种选择优势补偿了纯合患者死亡所失去的隐性基因(HbsHbs),因而维持了群体中的平衡多态性。类似的情况还有其它的有害基因,如地中海贫血、血红蛋白C、G6PD缺陷以及Duffy血型的Fy基因等。它们所以能在一定群体中维持较高的基因频率,就是因为有抗疟性使杂合子受到保护之故。
遗传负荷(geneticload)是指一个群体由于有害基因的存在而使其适合度降低的现象,有人描述其为整个群体遗传的无能性。因为致死基因是经突变产生的,可使生物在成年前而死亡,其基因既不能传于下一代,当然也就不利于生物个体的生存和延续后代。
遗传负荷一般用一个群体中每个个体带有的有害基因的数量来衡量。它包括突变负荷和分离负荷。突变负荷(mutation load)是一个群体中反复发生的突变产生了致死或亚致死基因。由于这些基因的积累,形成了选择上不利的纯合子,从而使群体平均适合度降低。分离负荷(segregation load)是指有害基因从有利的杂合子分离而产生了选择不利的纯合子,从而使群体由较高适合度的杂合子形成较低适合度的纯合子。遗传负荷有不同的估算方法。据估计,我国人群中至少每人有5-6个有害基因以杂合方式存在。
第三节 近亲婚配
1.近亲婚配和近交系数
近亲是指两个人在几代(3-4代)之内有着共同祖先,如果他们之间进行婚配就称为近亲婚配(consanguineous marriage,inbreeding)。在这种婚配情况下,由于夫妇双方都可能传来共同祖先的同一基因,而又可能把此同一基因传给他们的子女。这样,同一基因纯合概率会增加,所以近样结婚可导致常染色体隐性遗传病在后代中发病率增高的有害后果。
由于近亲婚配,在子女得到一对相同等位基因概率称为近交系数(近婚系数)(inbreeding coefficient)。
2.近亲系数的计算
(1)常染色体基因的近交系数:首先要说明和计算同胞兄妹间的近交系数。
1)同胞兄妹间的近交系数:设一对同胞兄妹的父亲有一对等位基因A1A2,母亲有一对等位基因A3A4(图6-1),他们子女中有A1A2、A2A3、A2A4基因型各1/4。从图可见,P1P2将这4个基因传至S,每代都是1/2机会,经过4步传递才可使S具有基因型A1A1、A2A2、A3A3或A4A4,所以4步传递的概率为(1/2)4。如果不论A1、A2、A3或A4,只看纯合子总概率,就是4×(1/2)4=1/4,即同胞兄妹间的近交系数。
2)舅甥女(或姑侄)间的近交系数:如图6-2所示,P1的基因A1经B1传给S需要2步,这样S基因型A1A1共需5步,其概率为(1/2)5,其它基因型A2A2.A3A3和A4A4也是一样,因此,舅甥女之间的近交系数为4×(1/2)5=1/8。
图6-1 同胞兄姝婚配中基因传递图解
图6-2舅甥女婚配中基因传递图解 C:甥女
3)表兄妹间的近交系数:此种情况较舅甥女婚配多了一步,如图6-3所示,其近交系数为4×(1/2)8=1/16。
4)二级表兄妹(从表兄妹)间的近交系数:由于这种情况较表兄妹婚配又多了2步,如图6-4所示,其近交系数为4×(1/2)8=1/64。
各种近亲关系和近交系数总结如表6-3。
表6-3 亲缘关系和近交系数
| 亲缘关系 | 近亲度 | 近交系数(F) |
| 父子 | 1 | 1/2 |
| 兄弟姐妹 | 1 | 1/2 |
| 同胞兄妹 | 2 | 1/4 |
| 舅甥女(姑侄) | 3 | 1/8 |
| 表兄妹 | 4 | 1/16 |
| 二级表兄妹 | 6 | 1/64 |
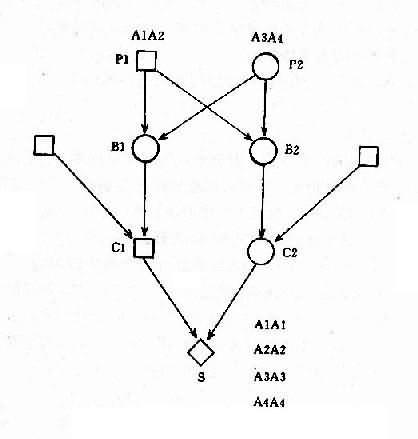
图6-3 表兄妹婚配中基因传递图解
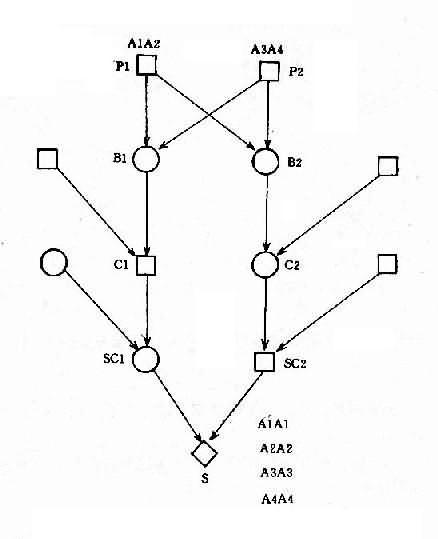
图6-4 二级表兄妹婚配中基因传递图解
(2)X连锁基因近亲系数:由于女性两条X染色体,有两个X等位基因,他们可形成纯合子,男性是半合子,不形成纯合子,对近亲婚配无影响。所以计算X连锁基因近交系数,只计算女儿的F值即可。而传递特点,男性x 连锁基因必定传给他的女儿,故其概率为1,反之,男性X连锁基因不传给他的儿子,故传递概率为0。这样,X连锁基因的近交系数与常染色体基因则不相同。
1)姨表兄妹的近交系数:如图6-5所示,P1的X1基因经B1.C1传至S只需计为1步(B1→C1),X1经B2.C2传至S计为2步(B2→C2)、(C2→S),所以,S为X1X1的概率为(1/2)3;P2之X2基因经B1、C1传至S计为2步,X2经B2、C2传至S计为3步,所以S为X2X2的概率为(1/2)5。同理,S为X3X3的概率也是(1/2)5。因此,姨表兄妹婚配的近交系数为(1/2)3+2×(1/2)5=3/16。
2)舅表兄妹的近亲系数:如图6-5所示,P1之X1基因传至B2时中断,故不能形成X2X2。基因X2从P2经B1、C1传至S计为2步,从X2从P2经B2、C2传至S,也只计2步,故
X2X2的概率为(1/2)4。同理,X3X3的概率为(1/2)4。因此,舅表兄妹婚配的近交系数为2×(1/2)4=1/8。
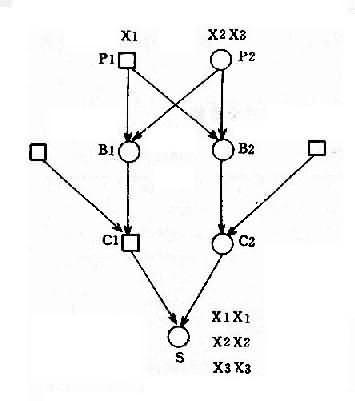
图6-5 姨表兄妹婚配中X连锁基因传递图解
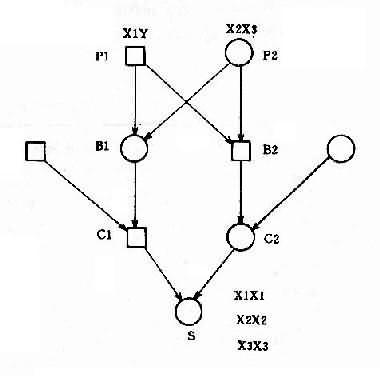
图6-6 舅表兄妹婚配中X连锁基因传递图解
3)如表兄妹、堂表兄妹的近交系数:如图6-7和6-8所示,基因X1从P1→B1传递中断,基因X2、X3从P2经B1至C1传递中断,所以其近交系数F值为0。因此,从上图的X连锁基因传递来看,姨表兄妹,舅表兄妹的婚配要比姑表兄妹和堂兄妹的婚配危害要大。
图6-7姑表兄妹婚配中X连锁基因传递图解
3.平均近交系数
即从群体角度来估计近亲婚配的危害,要计算平均近交系数(average inbreeding coefficient,a)。a值可按下列公式求出:
a=Σ(Mi/N)(Ⅰi)
Mi=某一类型近亲婚配数;N=总婚配数,Ⅰi=某一类型近亲婚配的近交系数。知道a值就可比较不同群体中近亲婚配的严重程度。一般来说,a值达1%左右已相当高。群体小,尤其是隔离群,a值较高,而大群体中,a值都很低。表6-4示一些群体的近亲结婚率和平均近交系数。
图6-8 姑表兄妹婚配中X连锁基因传递图解
表6-4 一些群体的近亲结婚率和平均近交系数
| 群体 | 近亲结婚率(%) | 平均近交系数 |
| 美国 | 0.11 | 0.00008 |
| 日本 | 8.16 | 0.004 |
| 法国 | 0.67 | 0.00023 |
| 德国 | 0.59 | 0.00019 |
| 意大利 | 1.90 | 0.00070 |
| 南印度 | 39.37 | 0.02834 |
| 埃及(Nubic) | 75.76 | 0.03335 |
| 中国汉族(北京、湖北) | 1.40 | 0.000665 |
| 回族(甘萧) | 9.7 | 0.00494 |
| 彝族(四川) | 14.6 | 0.00913 |
| 朝鲜族(克林) |
近亲婚配的危害主要表现为隐性遗传病纯合子患者的频率增高。以表兄妹婚配为例,他们所生子女是纯合子(aa)有两种原因:①近亲婚配,由共同祖先而来;②是由不同祖先分别而来。在①情况下,F值为1/16,a基因频率为q,这一途径形成纯合子aa的概率为1/16q,在②情况下,纯合子aa的概率为1(1/16)q2,他们子女中隐性纯合子概率为①+②即(1/16)p+(15/16)q2=q/16(1+15q)=q/16(p+q+15q)=(pq/16)+q2。而随机婚配中,所生子女的纯合子的频率为q2,与前者之比为(q2+pq/16])/q2。由此表明表兄妹婚配的子女隐性纯合子的频率增高pq/16。这种有害效应的大小与隐性基因频率(q)有关。如表6-5所示,q为0.10时,q2为0.01,表亲婚配后出生隐性纯合子的频率为0.015625,比随机婚配所出生的隐性纯合子频率高0.005625,两者之比为1.56。这表明大约3/5的纯合子来自表亲婚配,2/5的纯合子来自随机婚配。同理,q为0.01是两种不同婚配出生的纯合子之比为7.19,则表明大约7/8的纯合子来自表亲婚配,1/8的纯合子来自随机婚配。当q为0.001时,两者之比为63.5,更明显看出98.5%的纯合子来自表亲婚配,而1.5%的才来自随机婚配。因此,隐性遗传病越是少见,来自近亲婚配的患儿概率就越大。
表6-5 表婚配和随机婚配出生隐性纯合子的频率
| 基因频率(q) | 随机婚配→隐性纯合子频率(q2) | 表亲婚配→隐性纯合子频率(q2+pq/16) | pq/16 | 两者之比 |
| 0.2 | 0.04 | 0.05 | 0.01 | 1.25 |
| 0.10 | 0.01 | 0.15625 | 0.005625 | 1.56 |
| 0.04 | 0.0016 | 0.004 | 0.0024 | 2.5 |
| 0.02 | 0.0004 | 0.001625 | 0.001225 | 4.06 |
| 0.01 | 0.0001 | 0.000719 | 0.000619 | 7.19 |
| 0.001 | 0.000001 | 0.0000635 | 0.0000625 | 63.5 |
第四节 遗传漂变
由于某种机会,某一等位基因频率的群体(尤其是在小群体)中出现世代传递的波动现象称为遗传漂变(genetic drift),也称为随机遗传漂变(random genetics drift)。这种波动变化导致某些等位基因的消失,另一些等位基因的固定,从而改变了群体的遗传结构。在大群体中,不同基因型个体所生子女数的波动,对基因频率不会有明显影响。小群体的人数少,并与总人群相隔离,这种社会和地理因素形成的小群体,A基因固定(A=1),而a基因人很少,a基因的人如无子女,则a基因就会较快在人群中消失,造成此小群体中基因频率的随机波动。这种漂变与群体大小有关,群体越小,漂变速度越快,甚至1-2代就造成某个基因的固定和另一基因的消失而改变其遗传结构,而大群体漂变则慢,可随机达到遗传平衡。
一些异常基因频率在小隔离群体中特别高,可能是由于该群体中中少数始祖所具有的基因,由于遗传漂变而逐渐达到较高水平,这种现象称为建立者效应(founder effect)。例如,太平洋的东卡罗林岛中有5%的人患先天性色盲。据调查,在18世纪末,因台风侵袭,岛上只剩30人,由他们繁殖成今天1600余人的小群体,这5%的色盲,可能只是最初30人建立者的某一个人是携带者,其基因频率q=1/60=0.016,经若干世代的隔离繁殖,q很快上升至0.22,这就是建立者效应。
第五节 迁移
迁移或称移居(migration)是指具有某一基因频率群体的一部分,因某种原因移至基因频率不同的另一群体,并杂交定居,从而改变了群体的基因频率,这种影响也称迁移压力(migration pressure)。迁移压力的增强可使某些基因从一个群体有效地散布到另一群体中。大规模的迁移会形成强烈的迁移压力引起群体遗传结构的改变。与遗传漂变相反,这时,基因横跨种群障碍而慢慢扩散,造成一个大群体的基因频率逐渐改变,此过程称为基因流(gene flow),意为大群体间,由于迁移而造成基因交流,使群体间的基因差异逐渐消失。基因差异消失的快慢,取决于移居群体与接受群体间该基因频率的差异和每代移入基因的比例。小群体移入大群体影响小,大群体移入小群体影响大。对ABO血型不同等位基因频率在世界群体中分布的调查,提供了基因流一种很好的例证。ABO血型的B等位基因频率从东亚的0.30降至西欧的0.06,就是由于B基因在东方的原始突变后逐渐扩散到更多的西欧群体中。从东亚到西欧,由20%-30%逐渐下降至10%-20%、10%-15%、5%-10%、0-5%。又如苯硫脲的味盲者频率及其基因频率在欧洲及西亚白种人高,而在我国汉族中低,但在我国宁夏甘肃一带回族聚居的人群中处于两者之间。这可能是西亚波斯人在唐代经丝绸之路到当时长安进行贸易,以后又在附近定居,与汉族通婚,逐渐形成的一个群体。
第七章 基因定位
基因组是生物的生殖细胞中所含全部基因的总和。人类基因组具有极其复杂的结构,其编码蛋白质的结构基因大约有100 000个,每个单倍体DNA含有3.2×109bp,分布在24条常染色体和X,Y性染色体上。此外,还含有大量的非编码的重复DNA序列。基因定位(gene location)是用一定的方法将基因确定到染色体的实际位置。这是现代遗传学的重要研究内容之一。将不同的基因确定于染色体的具体位置之后,即可绘制出基因图(gene map)。
有两种基本方式制作人类染色体的基因图:即物理作图和遗传作图。物理作图(physical mapping)是从DNA分子水平制作基因图。它表示不同基因(包括遗传标记)在染色体上的实际距离,是以碱基对为衡量标准,所以物理图谱(physical map)最终是以精确的DNA碱基对顺序来表达,从而说明基因的DNA分子结构。从细胞遗传学水平,用染色体显带等技术在光学显微镜下观察,将基因定位不同染色体的具体区带,又称区域定位(regiona assignmer),而把基因只定位到某条染色体上称为染色体定位(chromosomalassignment)。这个水平上的基因图谱又称细胞遗传图(cytogenetical map)。分辨率可达5Mb至1Mb。遗传作图(geneticmapping)是以研究家族的减数分裂,以了解两个基因分离趋势为基础来绘制基因座位间的距离,它表明基因之间连锁关系和相对距离,并以重组率来计算和表示,以厘摩(cM)为单位。两个遗传座位间1%的重组率即为1厘摩。人类精细的遗传图水平可达1cM即100kb(1Mb)左右。
根据系谱分析和少数血型分析,就可确定某些基因位于X染色体上。X连锁关系确定后,再依重组率计算两者之间的相对距离,便可将两个基因定位于XX染体的相对位置上,1961年红绿色盲就是通过这种方法定位于X染色体上。
1968年Donahue依据系谱分析的原理,第一次将Duffy血型基因定位于第1号常染色体上。它在中期染色体时,发现1号染色体长臂1区的异染色质区有变异特征。继后,他发现在一些家族中,凡是具有这种形态特征的染色体都是Duffy血型者,从而将此血型的基因定位于第1号染色体上。伴随分子生物学和细胞分子遗传学的进展,基因定位的新方法不断出现,特别是体细胞杂交、分子杂交、DNA重组和DNA体外扩增(PCR)等技术后出现与应用,产生了许多定位新技术,如脉冲场凝胶电泳、染色体显微摄影、染色体步移和酵母人工染色体(YAC)克隆等,使基因定位的研究工作得到迅速发展。
自1973年第一次国际人类基因组制图(human genome mapping,HGM)会议召开以来,每隔2-3年就举行一次会议。在第一次HGM会议上,人类基因定位只有31个,而今迅速进展到已定位的基因4000多个,而且有一大批克隆基因和DNA遗传标记被定位,如表7-1所示。这些成果有力地推动了遗传咨询、基因诊断和基因治疗的进展,对医学理论和临床实践的发展起着十分重要的作用。
表7-1 HGM会议定位的克隆基因和DNA多态标记
| HGM11(1999年) | CCM(1992年) | HGM12/CCM93(1993年) | |
| 基因总数 克隆基因 多态标记 | 2778 1524 622 | 3301 713 | 4183 3808 850 |
| DNA节段总数 多态标记 克隆DNA | 6974 2608 9760 | 12376 3250 16277 | 25640 5114 29833 |
| 多态标记总数 微卫星 | 3230 312 | 3963 812 | 5964 2427 |
第一节 基因定位的方法
基因定位可从家系分析、细胞、染色体和分子水平进行研究,同时由于使用手段的不同派生出多种方法,不同方法又可联合使用,互为补充。现仅就几个主要方法加以说明。
1.体细胞杂交法体细胞是生物体除生殖细胞外的所有细胞。将从身体分离的体细胞做组织培养进行遗传学研究的学科称为体细胞遗传学(somatic genetics)。体外培养细胞可人为控制或改变环境条件,并可建立细胞株,长期保存,进行各种正常和病理研究。与基因定位有关的是体细胞杂交(somatic cell hybridization)。细胞杂交又称细胞融合(cellfusion),是将来源不同的两种细胞融合成一个新细胞。大多数体细胞杂交是用人的细胞与小鼠、大鼠或仓鼠的体细胞(hybrid cell)进行杂交。这种新产生的融合细胞称为杂种细胞(hybridcell),含有双亲不同的染色体。杂种细胞有一个重要的特点是在其繁殖传代过程中出现保留啮齿类一方染色体而人类染色体则逐渐丢失,最后只剩一条或几条,其原因至今不明。这种仅保留少数甚至一条人染色体的杂种细胞正是进行基因连锁分析和基因定位的有用材料。由于人和鼠类细胞都有各自不同的生化和免疫学特征,Miller等运用体细胞杂交并结合杂种细胞的特征,证明杂种细胞的存活需要胸苷激酶(TK)。但凡含有人第17号染色体的杂种细胞都因有TK活性而存活,反之则死亡。从而推断TK基因定位于第17号染色体上(图7-1)。这是首例用细胞杂交法进行的基因定位。由此可见,研究基因定位时,由于有杂种细胞这一工具,只需要集中精力于某一条染色体上,就可找到某一基因座位。
7-1 体细胞杂交基因定位示意图
2.克隆嵌板法(clone panel method)是应用杂种细胞保留或丢失人染色体有时有重叠现象而设计的一种简便有用的基因定位方法。如表7-2所示,选择3个都保留有4条人染色体的杂种细胞克隆,但染色体各不相同。如果某一表型特征只出现于克隆A、C而不见于B,就可决定该特征的基因只能在3号染色体上,因为A、C克隆都有3号染色体,而B克隆则无,其它亦可同理判断这样3个克隆可对8条(23=8)染色体作出判断;如果是5个克隆(25=32)就可对人类22条常染色体和2条性染色体作出判断。例如半乳糖激酶(GK)、尿苷-磷酸激酶(UMPK)和氨基已糖苷酶A(HEXA)的基因就是用此法分别定位于人的17号、1号和15号染色体上的。此外,还可对杂种细胞进行特殊染色,来识别杂种细胞中人和鼠染色体,以助基因定位。
表7-2 克隆嵌板示意
| 杂种克隆 | 保留的人染色体 | |||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
| A | + | + | + | + | - | - | - | - |
| B | + | + | - | - | + | + | - | - |
| C | + | - | + | - | + | - | + | - |
在体细胞杂交基础上还发展了微细胞(micro cell)技术,即制备只含少数或一条人染色体的微细胞进行细胞杂交。也可将人中期染色体进行分离直接转移到鼠类细胞中,使人染色体在受体细胞中裂解变成短的DNA片段,并整合到受体细胞的基因组中。如两个基因同时整合进去,就证明它们的紧密连锁关系。
3.原位杂交和荧光原位杂交重组DNA技术的建立与分子杂交相结合,从分子水平研究基因定位,发展了一系列有效方法。例如原位杂交(in situ hybridization)就是分子杂交技术在基因定位中的应用,也是一种直接进行基因定位的方法。分子杂交的基本原理是碱基的互补配对,同源的DNA-DNA双链或DNA-RNA链在一定条件下能结合成双链,用放射性或非放射性物质标记的DNA或RNA分子作为探针,可探测到细胞基因组中的同源部分。19770-1978年首次将分子杂交法应用于基因定位,即用α及β珠蛋白基因的cDNA为探针,与各种不同的人/鼠杂种细胞进行杂交,再对DNA杂交情况进行分析,找出cDNA探针与人染色DNA顺序间的同源互补关系,从而将人α及β珠蛋白基因分别定位于第16号和第11号染色体上。
图7-2 用原位杂交法将胰岛素基因定位于11p15
原位杂交的特点是杂交在显微镜载玻片上中期染色体标本上进行。所谓原位即指标本上DNA原位变性,在利用放射性或非放射性标记的已知核酸探针杂交后,通过放射自显影或非放射性检测体系来检测染色体上特异DNA或RNA顺序,可用放射性颗粒在某条染色体的区带出现的最高频率或荧光的强弱来确定探针的位置,从而进行准确的基因定位。图7-2表明用此法将胰岛素基因定位于11p15。但原位杂交必须在已知探针的情况下方可进行,而在未知致病基因时,则无法进行基因定位。
放射性同位素标记核酸探针与染色体显带结合进行原位杂交仍有不少缺点和局限性。例如需要使用放射性同位素的特殊实验室,自显影曝光时间长,同位素标记的探针不易保存,放射污染不利健康,特别是高质量的中期染色体标本在细胞培养基础上难以得到,观察费时,对间期核的研究难以进行等。
荧光原位杂交(florescencein-situ hybridization,FISH)是一种非放射性原位杂交方法。用特殊荧光素标记核酸(DNA)探针,可在染色体、细胞和组织切片标本上进行DNA杂交,对检测细胞内DNA或RNA的特定序列存在与否最为有效。探针不是放射性的而是将荧光染料与抗体蛋白结合进行检测。它们具有高度亲和力,有与放射性探针相同或更高的分辩率。现已可用不同的荧光染料同时进行多重原位杂交,显示出不同的荧光色泽。这种多色FISH技术近年来发展迅速,已成为基因定位作图和医学诊断的重要手段。1992年运用这种策略已能在中期染色体和间期细胞同时检测7个探针。科学家们的目标是实现24种不同颜色来观察22条常染色体和X、Y染色体。荧光原位杂交法提高了杂交分辩率,可达100-200kb。此法降低应用于基因定位外,还有多种用途,它已日益发展成为代替常规细胞遗传学的检测和诊断方法,在此不多论述。
4.连锁分析基因定位的连锁分析是根据基因在染色体上呈直线排列,不同基因相互连锁成连锁群的原理,即应用被定位的基因与同一染色体上另一基因或遗传标记相连锁的特点进行定位。生殖细胞在减数分裂时发生交换,一对同源染色体上存在着两个相邻的基因座位,距离较远,发生交换的机会较多,则出现基因重组;若两者较近,重组机会较少。重组DNA和分子克隆技术的出现,发现了许多遗传标记——多态位点,利用某个拟定位的基因是否与某个遗传存在连锁关系,以及连锁的紧密程度就能将该基因定位到染色体的一定部位,使经典连锁方法获得新的广阔用途,成为人类基因定位的重要手段。
染色体上两个位点从亲代传给子代时,若相距1cM,就有1%的重组机会。整个人类基因组含3.2×109bp,相应约有3300cM,每个染色体平均约有150cM,1cM约为1000kb。因此,一个致病基因和标记位点紧密连锁,二者不须在同一条染色体的同一区段,一条染色体可以产生大量的DNA多态,只要提供足够的家系,按孟德尔方式遗传的疾病都可将其基因定位。
图7-3表示某一致病基因与一多态位点的关系。父(Ⅰ1)是显性遗传病患者,基因型为DN,他妻子(Ⅰ2)正常,基因型为NN,多态标记位点父为Aa,母为AA,在子代中,除Ⅱ4外,都是致病基因与多态标记位点的完全连锁,即都遗传了父亲的A基因和D基因,或是a基因和正常N基因。据此信息,就可确证父亲的致病基因是多态标记A基因连锁在同一染色体上,而Ⅱ4则是重组体,而具有标记基因A的个体并不都是患者,Ⅰ2就是这样,所以,在连锁分析中,多态标记是极为有用的。
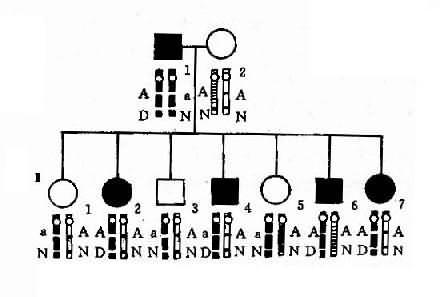
图7-3 致病基因与标记位点的边锁关系图
在人类基因组中还存在约50000-100000个串联重复序列家族,它们均匀穿插于基因组,平均50kb有一个,这些都是很好地遗传标记对突变的检测和基因定位研究起了重要作用。正如本章开始提到,至1993年10月已经发现多态标记总数为5964个,从分子水平进行基因定位,还有不少有效的新技术,它们相互配合使用,使基因定位得到迅速发展。此外,利用染色体自身结构及染色体畸变法进行缺失作图和基因剂量法等。总之,人们已不只是用单一技术进行基因定位,而是将细胞、染色体和分子水平技术结合起来,综合分析,互相印证,以达快速、准确的目的。
第二节 基因定位的应用
基因定位和基因图对遗传学、医学和人类及生物进化的研究都有十分重要的意义。它可提供遗传病和其他疾病的诊断的遗传信息,可以指导对这些疾病的致病基因的克隆和对病症病因的分析与认识,这些又取决于遗传图和物理图的相互依赖关系。通过多态位点标记进行连锁分析获得物理图的位置有助于遗传作图,同时通过连锁分析(部分有减数分裂的交换)又能指导物理作图,使基因定位更为精细。
1.连锁分析检测基因突变指导遗传病的诊断通过遗传连锁图可以在缺少任何生化或分子性质信息情况下对遗传病进行诊断。例如应用RFLP进行的临床诊断(详第十三章)。应当指出,由于多种遗传标记的定位,使RFLP的应用更加广泛。遗传图对疾病诊断的价值最明显的是当某一基因已被定位于某染色体,但尚未被克隆时,就可依赖于运用一个或多个遗传标记进行连锁分析,利用它和该基因的重组关系进行基因诊断。
2.连锁分析进行致病基因的鉴别与定位前段说明已知疾病基因与遗传标记相关进行连锁分析以诊断疾病,然而要认识这种关系就需要:①有足够数量家系来确定这种连锁;②有适当可提供信息DNA标记。后者较易,前者则难,尤其是稀少的疾病,患者在年幼时死亡。这时采取两种方法,一是少数的大家系,并从此家系成员中获得DNA信息,其优点是所有患者都是一个遗传病,并由单一基因突变引起;二是收集大量的小家系。前者如慢性进行性舞蹈病,后者如囊性纤维化(CF)。实际上,人们希望能发现连锁需在大约20cM距离或以下,再大的距离一般就难于发现连锁关系,特别是分子遗传学的新进展推出的反向遗传学――位置克隆的策略,更加速了疾病基因的鉴定与定位。
3.促进对癌基因和瘤抑制基因的定位与克隆,从慢性粒细胞白血病的Ph染色体的发现,了解到是染色体9和22的易位,以及Burkitt淋巴瘤染色体t(8:14)。1982年证实c-myc癌基因定位于8q24,并对其基因分子的结构有了深入认识。现今已知有100多种癌基因和约10种肿瘤抑制基因。而且肿瘤细胞遗传学异常的报道日益增多,到1993年10月,与染色体畸变相关的肿瘤总数达239种,其中结构畸变有188,数目畸变为51种。这些资料就充分说明基因定位和克隆对肿瘤的鉴别定位及其深入研究有着密切关系。当然,基因定位与基因图的发展,也推动了许多常见复杂病的遗传因素的研究。
4.位置克隆与基因定位基因定位的重要应用之一是据此进行基因克隆(gene cloning)。致病基因克隆有两种基本策略:一是功能克隆,二是位置克隆。
(1)功能克隆(funetional cloning):是利用疾病已知的遗传损伤而引起的生化功能如蛋白质基酸缺陷的信息,进行基因定位,进而克隆该致病基因。以进行的基因克隆大都是预先测知疾病基因的编码蛋白质,利用其mRNA反转录成cDNA,再用cDNA作探针,从人基因组中“约”出基因本身。然而,绝大多数遗传病的基因产物不明,就无法用功能克隆策略进行基因克隆。
(2)位置克隆(positional cloning):是先进行基因定位作图,然后找一来自该区的基因并进行克隆,在此之后才能明确该基因的功能。早先称之为逆向遗传学(reverse genetics),但究其基因鉴定到基因定位制图的全过程并非都是“逆向”的,而具遗传学的传统模式,故现多称其为位置克隆。
据HGM93国际会议的资料,至1993年,运用位置克隆策略克隆的人类疾病基因共有26个,其中1993年就有11个,如慢性进行性舞蹈病(HD)基因,肝豆状核变性(Wilson病,WND)基因等。位置克隆的操作过程如图7-4所示。
遗传病致病基因运用位置克隆的成功例子很多,典型之一是假肥大型肌营养不良(DMD)基因的克隆。学者们利用受累女性X染色体与常染色体21的易位,以及男患儿发生小的Xp21.2缺失并伴发3种其它X连锁隐性遗传病,再运用RFLP连锁分析将DMD基因定位于Xp21。Ray等利用了X与21号染色体基因易位,于1986年克隆了该基因的一部分,即XJ系列探针。Kunkel等于同年利用男患儿(B.B)的DNA,通过灵敏的技术提纯得自有Xp21.2缺失的片段,即后来称为pERT87系列的探针。虽然细胞遗传学检查未见此缺失,但患者的DNA中确有
图7-4 位置克隆操作过程图解
图7-5 pERT87系列探针对7个DMD患儿的Sorthern印迹分析
图中患儿1、7无缺失,2-6出现缺失,所有患儿均未见细胞遗传学的缺失
pERT87的缺失,这说明该片段可能是DMD基因的一部分。图7-5表示用DNA印迹杂交法,观察到pERT87区附加的缺失片段(87-1、87-2、87-18等),而且缺失片段不断向此克隆区两侧扩大,表明DMD可能是一个很大的基因。
为证明该区的DNA片段是否含有DMD基因的外显子,又用Northern印迹法检测一个16kb的特异肌肉转录物,接着克隆出它的cDNA,最后证明DMD基因的确很大,约为2300kb,占X染色体的1%以上,该基因编码肌营养不良蛋白(dystrophin),并测出它的氨基酸顺序,该蛋白影响纹肌和心肌的结构和收缩功能。
关于基因定位,近年又兴起一种候选基因方法(candidate gene approach)。此法无需经过基因图和物理图的过程,而根据某种遗传病和特定基因分别定位于染色体的同一区域,而后再发现并鉴定其基因突变,则可研究此致病基因。自1992-1993年用此法克隆基因已有十几个,如SOD1(肌萎缩性侧索硬化症)、RET(多发性内分泌促瘤2A型)基因等。
第三节 人类基因组计划
基因定位取得了巨大成就,新的分子生物学技术不断发展与完善,使许多学者认为已到了对人类基因组全部30亿bp,5-10万个基因进行整体制图和测序的时候,以达到人类基因组完善而系统认识的目的。1985年美国学者提出了人类基因组项目(human genome project),引起学术界巨大反响和热烈争论,经两次会议研究后,1988年美国国会批准,由国立卫生院(NIH)和能源署(DOE)负责执行,此后成立了国际性组织――HUGO(human genome organization),组织了国际性的合作研究。这是一项耗资数十亿美元,全球性的大科学项目,预计2005年可完成基因组顺序分析,这将对生命科学的发展作出巨大的理论和实用价值的贡献。
1990年美国又建立了国家研究中心和国内若干大的中心。1992年法国研究组构建了1-1Mb YAC文库,生产了大量的遗传标记,首先绘制出21号染色体长臂的重叠YAC克隆,引起了科学界的轰动。HUGO先后于1988-1993年每年召开一次人类基因组的国际会议。1992年在巴西还召开了第一次南北人类基因组会议,使发展中国家也参与了这项研究,并提出“人类遗传多样性项目(human genetic persity project),得到与会各国支持。我国与于1993年正式组织人类基因组研究,标志我国也正式参与了这基世界性宏伟科学研究工程。
人类基因组项目主要目标与程序:①研究人类遗传的基础结构;②确立人类生物学的DNA顺序;③进行基因的生化分析,也就是要进行人类基因定位。全部核苷酸顺序的分析,有助于了解结构,认识功能,亦即人类能够“读出”并“读懂”人类基因组的全部ATGC语言,从遗传学来认识人类正常功能和病理变化,也是从分子水平来认识人类自身的结构与功能特征。
人类基因组全顺序分析分两大步骤:即制图(mapping)和测序(sequencing),同时发展人类和模型生物基因定位和测序的新技术。全过程又可分为4个阶段(phases)如图7-6所示:①构建1cM的遗传图;②构建物理图;③建立重叠克隆系(cntig);④;完成核苷酸顺序测定。当然这4个阶段是相互交叉进行的。由于新技术的不断涌现,现今定位的基因与日俱增,许多DNA多态遗传标记的运用,已可制作相邻标记平均距离为5cM的基因或DNA片段,其中有的已达1-2cM。1995年可制出1-5cM的遗传图。物理图的制作在用FISH技术和YAC库后,已可制出多色细胞遗传图谱,分辩率可达100-200kb。全自动化测序仪不断更新,极大提高了测序的准确性和效率。
图7-6 人类基因组研究阶段示意图
A.1cM的基因图构建;B。物理图构建;
C.建立重叠序列测定;D核苷酸序列测定
人类基因组项目的研究重要内容包括致病基因的鉴定、克隆与分析(图7-7)。这不仅局限于孟德尔式的单基因遗传病,还包括人类肿瘤和众多常见复杂病等。研究的成果不仅使人们从分子水平阐明各种疾病发生的机理,同时也认识正常生物学结构和功能,其最终成果必将是巨大的,甚至尚难以预料所产生的深远影响,但至少可以看出,它将促进从整体到细胞水平全面深入发展为分子医学,并揭示人类自身的奥秘,用遗传学语言来阐明生命的本质和各种生命现象。
图7-7 7号染色体的基因图示
7号染色体上已定位的遗传病,不包括已定位的DNA片段及其他未导致遗传病的基因
第八章 药物遗传学
药物遗传学(pharmacogenetics)是生化遗传学的一个分支学科,它研究遗传因素对药物代谢动力学的影响,尤其是在发生异常药物反应中的作用。
临床医生在使用某些药物时,必须遵循因人而异的用药原则。因为在群体中,不同个体对某一药物可能产生不同的反应,甚至可能出现严重的不良副作用,这种现象称为个体对药物的特应性(idiosyncracy)。特应性产生的原因相当部分取决于个体的遗传背景。
众所周知,药物在体内要经过吸收、分布、代谢和排泄,才能完成药物发挥药效的过程。在此过程中,许多环节都与酶和受体的作用密切相关。倘若决定这些酶或受体蛋白的基因出现变异或缺陷,必将导致药物代谢发生异常反应。因此,有必要深入了解遗传变异对药物反应的影响及其分子基础,并据此预测对药物异常反应的个体,从而进行有效的防治。对药物遗传学的研究,已揭示了许多药物异常反应的遗传基础和生化本质,这对于指导临床医生正确掌握用药的个体化原则,防止各种与遗传有关的药物反应都具有指导价值。
第一节 药物反应的遗传基础
一、琥珀酰胆碱敏感性
琥珀酰胆碱(succinylcholine,suxamethonium)是一种肌肉松弛剂,早期作为外科麻醉剂使用,它不仅可使一般骨骼肌松弛,而且可使呼吸肌短暂麻痹(2-3分钟),但有极少数人(1/2000)在用药后呼吸停止可持续一小时以上,如不行人工呼吸,往往导致死亡。但若立即输血,呼吸可很快恢复。后来知道,琥珀酰胆碱在血中可被血浆中假胆碱酯酶(pseudocholinesterase,简称酯酶)水解而解毒,故作用短暂。琥珀酰胆碱敏感者,血浆酯酶活性缺乏或缺如,使琥珀酰胆碱作用时间延长,以致中毒。
现知琥珀酰胆碱敏感性是属常染色体隐性遗传,控制酯酶的基因为E1和E2。已发现的变异型有5种:E1a、E1a、Ef1、E1s、E+2及E2cynthiana。其中仅纯合子E1sE1s酯酶活性最低(酶活性0-5%),较常见的E1aE1a型酶活性也低35%。
二、异烟肼慢灭活
异烟肼(isoniazid)是常用的抗结核药。在体内主要通过N-乙酰基转移酶(N-acetyltransferase,简称乙酰化酶)。将异烟肼转变为乙酰化异烟肼而灭活(图8-1)。按对异烟肼灭活的快慢,人群中可分出两类:一类称为快灭活者(rapid inactivator),血中异烟肼半减期为45-110分钟;另一类称为慢灭活者(slow inactivator),半减期2-4.5小时。而慢灭活者是由于乙酰化酶的遗传缺乏故灭活较慢。此酶系由常染色体一对等位基因控制。快灭活者(RR)与慢灭活者(rr)均为纯合子,杂合子(Rr)则具有中等乙酰化速度,不同种族慢灭活者发生率不同:埃及人高达83%,白种人50%左右,黄种人10%-30%,爱斯基摩人仅为5%。由于异烟肼乙酰化+速度的个体差异对结核病疗效有一定影响。如每周服药1-2次则快灭活者疗效较差。但从毒性作用看,慢灭活者有80%发生多发性神经炎(polyneuritis),而快灭活者仅20%有此副作用。这是由于异烟肼在体内可与维生素B6反应,使后者失活,从而导致B6缺乏性神经损害,故一般服异烟肼需同时服用B6可消除此种副作用。此外,服用异烟肼后有个别人可发生肝炎,甚至肝坏死。发生肝损害者中86%是快灭活者,其原因是,乙酰化异烟肼在肝中可水解为异烟酸和乙酰肼,后者对肝有毒性作用。

图8-1 异烟肼的灭活过程
通过N-乙酰基转移酶进行乙酰化灭活的药物尚有磺胺二甲嘧啶、苯乙肼、普鲁卡因酰胺、甲基硫氧嘧啶、肼苯达嗪、氨苯砜等。对这些药物快慢灭活的临床意义仅有一些零星材料,尚需进一步探讨。
三、葡萄糖-6-磷酸脱氢酶缺乏症
葡萄糖-6-磷酸脱氢酶(glucose-6-phosphate dehydrogenase,G6PD)缺乏症是一种主要表现为溶血性贫血的遗传病,一般平时无症状,但在吃蚕豆或伯氨喹啉类药物后出现血红蛋白尿、黄疸、贫血等急性溶血反应。
众所周知,红细胞中糖代谢主要是通过无氧糖酵解进行,但也有10%通过戊糖代谢旁路(图8-2)。G6PD活性正常是,可以生成足量NAKPH,从而保证了红细胞中GSH含量。GSH通过下列反应可消除机体在氧化还原过程中(特别是气体性药物作用下)生成的H2O2的毒性作用。

不难看出,若G6PD缺乏,NADPH生成不足,则红细胞GSH含量减少,H2O2可迅速将GSH破坏,过多的H2O2氧化Hbβ链表面半胱氨酸的SH基。表面SH氧化后,Hb的4条肽链接触面不稳定而散开,Hb内部的SH也被氧化,导致Hb变性。变性的珠蛋白附着于红细胞膜上,在显微镜下可观察到,即变性珠蛋白小体(Heinz小体)。此外,H2O2还可氧化红细胞膜上的SH基,故这种红细胞易在血中破坏。最近研究表明,NADPH的减少本身,也降低了红细胞对H2O2的抵抗作用。由于以上原因,红细胞变形性降低,不易通过脾(或肝)窦而遭阻留破坏,引起血管内和血管外溶血。
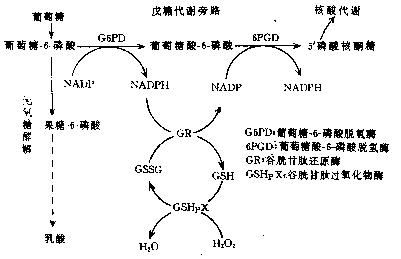
图8-2 红细胞的戊糖代谢旁路
基因突变所产生G6PD生化变异型已报告400种以上。中国人中已发现30多种。根据临床表现可分为二类:①酶活性严重缺乏(酶活性测不出)伴有非代偿性慢性溶血(属非球形溶血性贫血);②酶活性严重缺乏(活性<10%)或中度缺乏(10%-60%),仅在有诱因作用后才溶血,我国多为后一类。也有酶活性轻度降低、正常(60%-150%)或升高(>150%)的变异型,一般无溶血现象。
图8-3 G6PD缺乏症女性杂合子的外周血片示嵌合体,细箭头示正常红细胞;粗箭头示G6PD缺乏红细胞
G6PD基因定位于Xq28,由13个外显子组成,全长18kb,编码515个氨基酸。G6PD缺乏症呈X连锁不完全显性遗传,男性半合子呈显著缺乏,女性杂合子酶活性变异范围大,可接近正常亦可显著缺乏。根据Lyon假说,女性杂合子实际上应是含有G6PD缺乏红细胞和正常红细胞的嵌合体,这已从形态学上证实(图8-3),两种细胞系的细胞嵌合数量不同直接影响女性(G6PD)缺乏杂合子的酶活性水平,故在临床上具有不同的表现度。
由于生化变异型已报告太多,靠酸蛋白的生化学特点(如电泳速率,热稳定性等)来区分出新的变异型已很困难。自1986年克隆了此基因,特别是1991年发表了此基因的DNA全顺序后,就已从DNA水平鉴定G6PD基因的突变类型。目前已知,G6PD基因的主要突变形式是点突变。国际上报告了50多种点突变和1种1个密码子缺失的缺失型突变,其中中国人已报告11种点突变(表8-1)。不同生化变异型可以具有相同的点突变;也有不同点突变具有同一生化异型。有的突变只产生多态性而与酶活性降低无关。
表8-1 中国G6PD缺乏者中的11种点突变
| 类型 | 顺序(cDNA) | 碱基改变 | 氨基酸置换 |
| Ch1 | 1376 | G→T | 精→亮 |
| Ch2 | 1388 | G→T | 精→组 |
| Ch3 | 1311 | G→T | 无 |
| Ch4 | 392 | G→T | 甘→缬 |
| Ch5 | 1024 | G→T | 亮→苯丙 |
| Ch6 | 95 | A→G | 精→组 |
| Ch7 | 592 | C→T | 精→半胱 |
| ″Chinese1″ | 835 | A→T | 苏→丝 |
| ″Chinese2″ | 1360 | C→T | 精→半胱 |
| ″Chinese3″ | 493 | A→G | 冬酰→天冬 |
| CT2 | 487 | G→A | 甘→丝 |
注:Ch3的内含子Ⅺ93位有C→T突变。Ch3突变为为一种同义突变,具有多态特点
G6PD缺乏症呈世界性分布,但比较集中于亚热带地区。据估计全球G6PD缺乏症患者达2亿人。我国主要分布于黄河流域以南各省,尤以广东、广西、海南、贵州、云南、四川发生率高,约4%-20%。
G6PD缺乏症是某些常见药物性溶血的遗传基础。已知能引起G6PD缺乏者溶血的药物和化学制剂有数十种之多,有不少是常用药物(图8-2)。故G6PD缺乏症患者因治疗必须使用表8-2中某些药物时,应在医生严密监护下使用。G6PD缺乏症尚是蚕豆病、新生儿黄疸、某些感染性溶血(如病毒性肝炎、流感、大叶性肺炎、伤寒、腮腺炎等)发生的遗传背景,其中新生儿黄疸引起核黄疸可导致患儿智力低下,甚至死亡。
表8-2 G6PD缺乏者应禁用或慎用的药和、化学制剂及食物
| 抗疟药:伯氨喹啉,扑疟母星,氯喹 |
| 磺胺药:磺胺,乙酰横胺,横胺吡啶,TMP-SMZ等 |
| 砜类药:氨苯砜,普洛明 |
| 止痛药:阿司匹林,非那西丁 |
| 杀虫药:β萘酚,锑波芬,来锐达唑(nitridazole) |
| 抗菌药:硝基呋喃类,氯霉素,对氨水杨酸 |
| 共它:蚕豆,丙横舒,BAL,大量维生素K等 |
四、血卟啉症
药物反应也可以在遗传病的基础上发生。血卟啉症(porphyrias)的急性发作就是一个典型的例证。血卟啉症是一组涉及血红素合成有关酶遗传性缺陷的疾病,除其中有些类型表现为对光敏感皮肤出现红斑、水疱、溃疡、感染等症状外,其余常见类型主要表现为急性腹痛、便秘、呕吐、周围神经运动障碍(如肌无力、麻痹)以及精神症状(幻觉、精神错乱、焦虑等),尿中和粪中卟啉及卟啉前体物质增多。不同酶缺乏是形成各种类型的遗传基础。病人的缓解期可无症状,仅有尿和粪中卟啉类物质排泄增多。但多种药物,可诱导急性发作,如巴比妥、利眠宁、眠尔通、磺胺类药、苯妥英钠、灰黄霉素、雌激素等。药物诱发本病发作的机理未明。有人认为,这些药物可以加速δ氨基γ酮戊酸(ALA)合成酶的合成。ALA增多因而生成的胆色素原(尿和粪卟啉前体物)也增多。预防本病发作的有效方法就是避免使用上述药物及其他诱发因素(如饮酒、日晒等)。
其他具有遗传基础的药物反应及其表现列于8-3。
表8-3 其他具有遗传基础的药物反应
| 症状或病名 | 诱发因素 | 异常的酶或蛋白质 | 临床表现 |
| 无过氧化氢酶轿症 | H2O2 | 这氧化氢酶 | 髟过氧化消毒无反应 |
| 糖尿病 | 氯磺苯脲 | ? | 饮洒后充血面赤 |
| 苄丙酮香豆素耐受性 | 苄丙酮香豆素 | 维生素K环氧化物还原酶 | 抗凝后充血面赤 |
| 青光眼 | 糖皮激素 | ? | 眼压增高 |
| 痛风 | 氯噻嗪 | ? | 关节肿痛 |
| 不稳定血红蛋白病 | 磺胺类、氧化剂 | 不稳定血红蛋白 | 溶血性贫血 |
| 恶性体温过高 | 麻醉剂 | ?(肌浆钙蛋白) | 体温过高 |
| 高铁血红蛋白还原酶 | 亚硝酸盐,氧化剂 | NADH-高铁血红 | 紫绀,高铁血红蛋白 |
| 缺乏症 | 蛋白还原酶 | 血症 | |
| 周期性麻痹 | 胰岛素、肾上腺素等 | ? | 瘫痪 |
第二节 毒物反应的遗传基础
有时,药物和毒物并没有严格的界限,药物过量可引起中毒,少量毒物有时也可作为药物使用,特别是环境中某些诱变剂、致癌剂或致畸剂在群体中引起不同的个体反应,某些个体对这些有害因子表现易感倾向。实际上环境中各种有害因子在人体内的代谢途径也可能受特定基因型的制约,药物遗传学的某些原则,也可应用于研究毒物对不同基因型的中毒效应。生态遗传学(ecogenetics)是研究群体中不同的基因型对各种环境因子的特殊反应形式,因此,毒物和药物遗传学都可归入生态遗传学的研究范围。现举2例说明毒物中毒个体差异的机理。
一、酒精中毒
人们早已观察到,人类对酒精耐受性有种族的和个体的差异。酒精敏感者,当摄入0.3-.05ml/kg体重乙醇时,即可表现面赤、皮湿升高、脉率加快等酒精中毒症状,而酒精耐受者则否。黄种人中80%为敏感者,白种人中仅5%敏感者。
现知,酒精在体内的代谢过程主要由肝中的乙醇脱氢酶(alcohol dehydrogenase,ADH)和乙醛脱氢酶(aldehydedehydrogenase,ALDH)所制约,如下式:
C2H5OH+NAD+[ ]CH3CHO+NADH+H+
CH3CHO+NAK+H2O[ ]CH3CHO+NADH+H+
在第一个反应生成的乙醛可刺激肾上腺素、去甲肾上腺素等物质的分泌,引起面红耳赤、心率快、皮温高等症状。
ADH是二聚体,由3种亚单位α、β、γ组成,α、β、γ分别由ADH1、ADH2、ADH3基因编码。成人主要是β链二聚体。ADH2具有多态性,大多数白种人为ADH21,由β1β1组成;而90%黄种人为ADH22由β1的变异肽链β2(47位半胱氨酸→组氨酸)组成(β2β2)。β2β2的酶活性约为β1β1的100倍,故大多数白种人在饮洒后产生乙醛较慢,而黄种人积蓄乙醛速度较快。
ALDH亦有2种同工酶:ALDH1和ALDH2。黄种人中有3种表型:①普通型,ALDH1与ALDH2均有;②常见的“非经典型”,仅有ALDH1无ALDH2占50%;③罕见“非经典型”,仅有ALDH2无ALDH1,只在个别日本人中发现。几乎所有的白种人都为普通型。ALDH2活性较ALDH1活性高。
具有ADH2及ALDH1者对酒精最敏感;具有ADH1及ALDH1者次之;具有ADH2及ALDH2者最不敏感,这是黄种人较白种人易产生酒精中毒的原因。
综上所述,黄种人较白种人易产生酒精中毒的原因是遗传因素决定的。大多数黄种人在饮酒后产生乙醛速度快,而氧化为乙酸的速度慢,故易产生乙醛蓄积中毒。
二、吸烟与慢性阻塞性肺疾患
慢性支气管炎(chronicbronchitis)合并肺气肿(emphysema)或支气管哮喘(bronchial asthma)统称为慢性阻塞性肺疾患(chronicobstructive pulmonary disease,COPD)。这类疾病以肺泡破坏、融合、呼吸面减少导致缺氧甚至肺心病为特征。大量调查表明,COPD的发生与吸烟有关。这是由于吸烟刺激巨噬细胞和中性粒细胞释放弹性蛋白酶,从而分解肺弹性蛋白之故,但远非吸烟者都患COPD。现在知道,COPD的产生也有一定的遗传基础。
正常人机体内存在一组能抑制蛋白酶活性的多肽和蛋白质,称为蛋白抑制物(proteinase inhibitors),广泛分布于各种组织,以内分泌腺和血浆中含量最高。α1抗胰酶(α1-antirtypsin,α1AT)是血清中主要的蛋白酶抑制因子,它能抑制多种蛋白酶(包括弹性蛋白酶)活性。
α1AT基因位于染色体14q32.1,全长10226bp,含有5个外显子,编码394个氨基酸。α1AT是一种急性反应蛋白,主要功能为保护组织免受蛋白酶的分解。正常α1AT的反应中心处于暴露位置,当与蛋白酶作用时,第358位甲硫氨酸(反应中心)与359位丝氨酸之间的肽链发生断裂,环状结构被破坏,与蛋白酶形成一稳定的复合物,从而使其失去活性。
血清电泳表明,α1AT存在遗传多态性,至少有33种变异型。每种变异型对蛋白酶抑制活性都不相同(图8-4)。Pim 是世界上各种人群里最常见的等位基因,基因频率均介于0.866-0.996;而PiS为0.11-0.12;Pizz最低,仅为0.01-0.002。SS型为MM型羧基端第131位谷氨酸被缬氨酸所取代(T→A);ZZ型为羧基端第53位谷氨酸被氨酸所取代(C→T)。调查表明,COPD患者中ZZ型频率较正常人中的ZZ型频率高43倍。ZZ型者由于缺乏α1AT活性,吸烟诱导产生的弹性蛋白不能受到有效的抑制,从而“消化”肺,泡导致COPD产生。

图8-4常见α1AT变异型电泳图谱及其活性
此外,还已证明ZZ型者易患新生儿肝炎、不明原因的肝硬化及慢性活动性肝炎。例如新生儿肝炎中20%为ZZ型婴儿,原因不明。
三、吸烟与肺癌
众所周知,吸烟者易患肺癌,但远非所有嗜烟者均患肺癌,有证据表明,吸烟者是否患肺癌与个体的遗传基础可能有关。
烟叶中含有致癌的多环苯蒽化合物,但致癌癌性较弱,进入机体后通过细胞微粒体中芳烃羟化酶(aryl hydrocarbonhydroxylase, AHH)的作用可转变为具有较高致癌活性的致癌环氧化物。此外,苯蒽化合物还有诱导AHH活性作用。其诱导作用的高低因人而异,受遗传因素决定。人群中可区分为高诱导组,中等诱导组和低诱导组。据调查:如果以低诱导组发生肺癌的易感性为1,中诱导组为16倍,高诱导组则高达36倍。
第九章 肿瘤与遗传
在人们生活的环境中存着不少物理的、化学的和生物的致癌因子,它们在一定条件下可以诱发肿瘤。例如,各种电离辐射和紫外线照射可以引起白血病和皮肤癌;多环芳烃化合物如3,4-苯并芘可以引起肺癌,黄曲霉素可以诱发肝癌;亚硝受可以引起各种消化道肿瘤;在生物因子中,已经证明某些病毒可以引起动物肿瘤,并与一些人类肿瘤如鼻咽癌、白血病密切有关。这些因子是通过引起基因异常而致瘤的。
但是尽管人们都接触各种致癌因子,却远非人人都发生肿瘤,这表明还存在个体的易感性,而易感性在很大程度是遗传物质的结构或功能才能使正常细胞转变为癌细胞,但对不同肿瘤,环境因素只有改变遗传因素作用的大小各异。
近年来肿瘤的分子遗传学研究表明,一些与细胞的生长和分化有关的基因在癌变过程中起关键作用,这些基因称为癌基因和肿瘤抑制基因,它们的结构或功能异常使细胞得以无控制生长,并最终导致肿瘤发生。
因此,有人认为,肿瘤是一种遗传病,可称为体细胞遗传病,因在其发生发中基因及基因异常起着重要作用。已知一些肿瘤是按照孟德尔方式遗传的,而在另一些肿瘤中遗传的“易感基因”和环境因素共同发挥作用;还有一些肿瘤是由于特定基因发生体细胞突变引起的,这种突变虽然不是遗传得来的,但却发生在遗传物质。
道德列举一些事实说明肿瘤发生中的遗传因素,然后再介绍瘤细胞的染色体和基因异常以及肿瘤发生的遗传机理。
第一节 肿瘤发生中的遗传因素
一、肿瘤的家族聚集现象
1.癌家族癌家族(cancerfamily)是指一个家系中恶性肿瘤的发病率高(约20%),发病年龄较早,通常按常染色体显性方式遗传,以及某些肿瘤(如腺癌)发病率很高等。Lynch将上述特点归纳为“癌家族综合征”。曾经报告过一个癌家族(G家族,图9-1),经地70多年(1895年开始)间的五次调查,有些支系已传至第七代,在842名后裔中共发现95名癌患者,其中患结肠腺癌(48人)和子宫内膜腺癌(18人)者占多数。这95人中有13人肿瘤为多发性,19人癌发生于40岁之前;95名患者中72人有双亲之一患癌,男性与女性各47和48人,接近1:1,符合常染色体显性遗传。
图9-1 癌家族G部分系谱图
2.家族性癌家族性癌(familialcarcinoma)是指一个家族内多个成员患同一类型的肿瘤,例如,12%-25%的结肠癌患者有肠癌家族史。许多常见肿瘤(如乳腺癌、肠癌、胃癌等)通常是散发的,但一部分患者有明显的家族史。此外,患者的一级亲属中发病率通常高于一般人群3-4倍。这类癌的遗传方式虽然还不很清楚,但表明一些肿瘤家族聚集现象,或家族成员对这些肿瘤的易感性增高。
再者,在对77对患白血病的双生子调查中发现,同卵双生者发病一致率非常高;在另一调查中,20对同卵双生子均患同一部位的同样肿瘤。这些都说明遗传因素在肿瘤发病中的作用。
二、肿瘤发病率的种族差异
某些肿瘤的发病率在不同种族中有显着差异。如在新加破的中国人、马来人和印度人鼻咽癌发病率的比例为13.3:3:0.4.移居到美国的华人鼻咽癌的发病率也比美国白人高34倍。其它一些肿瘤类似情况。如黑人很少患Ewing骨瘤、睾丸癌、皮肤癌;日本妇女患乳腺癌比白人少,但松果体瘤却比其它民族多10余倍。种族差异主要是遗传差异,这也证明肿瘤发病中遗传因素起着重要作用。
三、遗传性肿瘤
一些肿瘤是按孟德尔方式遗传的,亦即由单个基因的异常决定的。它们通常以常染色体显性方式遗传,并有不同程度的恶变倾向,故也称为遗传性癌前改变。现举例如下。
1.家族性结肠息肉家族性结肠息肉(familialpolyposis coli,FPC)又称为家族性腺瘤样息肉症,在人群中的发病率为1:100000。表现为青少年时结肠和直肠已有多发性息肉,其中一些早晚将恶变。90%未经治疗的患者将死于结肠癌。FPC的基因现定位于5q21.
2.Ⅰ型神经纤维瘤Ⅰ型神经纤维瘤(neurofibromatosis,NF1)患者沿躯干的外周神经有多发的神经纤维瘤,皮肤上则可见多个浅棕色的“牛奶咖啡斑”,腋窝有广泛的雀斑,在少数患者肿瘤还有恶变倾向。现知与NF1发生密切有关的是一个肿瘤抑制基因,称为NF1基因,它定位于17q11.2,并已分离克隆。
此外,基底细胞痣综合征(basalcell nevus syndrome)、恶性黑素瘤(malignant melanoma)等属于遗传性肿瘤。
还有一些肿瘤既有遗传的,也有散发的。前者临床上按常染色体显性方式遗传,属遗传型,常为双侧性或多发性,发病早于散发型病例。这些肿瘤大多来源于神经或胚胎组织,虽然比较罕见,但在肿瘤病因研究中具有重要意义,故择要介绍如下。
1.视网膜母细胞瘤 视网膜母细胞瘤(retinoblastoma),为眼球视网膜的恶性肿瘤,多见于幼儿,大部分患者(70%)2岁前就诊,发病率为1:15000-28000。肿瘤的恶性程度很高,可随血循环转移,也能直接侵入颅内(图9-2)。
视网膜母细胞瘤可分为遗传型和散发型。大约40%的病例属遗传型,即由于父母患病或携带有突变基因,或父母的生殖细胞发生突变,在患儿出生时全身细胞已有一次视网膜母细胞瘤基因(Rb1)的突变。另约60%则是患者本人Rb1基因两次体细胞突变的结果,属非遗传型。遗传型患者常为双侧或多发肿瘤,平均发病年龄也较散发型者为早(15个月:30个月)。双侧性患者中还有少数患者可见一条13号染色体异常,主要是其长臂1区4带的缺失。13号长臂的这一区带正是视网膜母细胞瘤基因所在之处。
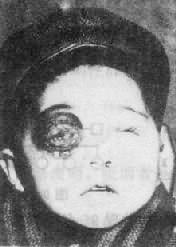
图9-2 双侧视网膜母细胞瘤右眼已萎缩
2.神经母细胞瘤 神经母细胞瘤(neuroblastoma)也是一种常见于儿童的恶性胚胎瘤,起源于神经嵴,活婴中的发病率为1:10000。神经母细胞瘤为常染色体显性遗传性肿瘤。有的神经母细胞瘤还合并有来源于神经嵴的其他肿瘤,如多发性神经纤维瘤、节神经瘤、嗜铬细胞瘤等。
3.Wilms瘤 Wilms瘤即肾母细胞瘤(nephr oblastoma),是一种婴幼儿肾的恶性胚胎性肿瘤,约占全部肾肿瘤6%,活婴中的发病率约为1:10000,3/4的肿瘤均在4岁以前发病。也可分为遗传型(38%)和非遗传型(62%),前者双侧性肿瘤较多,发病年龄较早,呈常染色体显性遗传,有明显的家族聚集现象。患者可伴有无虹膜症、半侧肥大、假两性畸形以及智力低下等。
一些易患Wilms瘤的无虹膜症患者有11号染色体短臂1区(11p13)缺失,而在Wilms瘤细胞中也曾发现11p13的缺失。现在认为11p13和11p15上有2个与肿瘤有关的基因,它们的异常都可能与Wilms瘤的发生有关。
四、染色体不稳定综合征
有一些隐性遗传病患者除易患肿瘤外,同时还有全身染色体容易断裂或对紫外线特别敏感的特点,这表明肿瘤与染色体不稳定之间有某种联系,这一类疾病称为染色体不稳定综合征,现择要介绍几种。
1.Fanconi贫血Fanconi贫血(Fanconi’sanemia)是一种儿童期的骨髓疾病,表现为全血细胞减少,故又称为先天性血细胞减少症(congentialpancytopenia)。患者有贫血、易疲乏、易出血和感染等症状,并常伴有先天畸形,尤其是大拇指或桡骨发育不良(或缺如),以及皮肤色素沉着等。患者的染色体自发断裂率明显增高,单体断裂、裂隙等染色单体畸变很多,双着丝粒体、断片、核内复制也很常见(图9-3)。约10%患者转变为白血病(主要是粒单型),且死于白血病者比正常人群高约20倍。
2.Bloom综合征Bloom综合征(Bloom’s syndrome)多见于东欧犹太人的后裔。患者身材矮小,对日光敏感,故面部常有微血管扩张性红斑。患者外周血培养细胞有各种类型的染色体畸变和单体畸变,包括许多对称的四射体,姐妹染色单体交换率也比正常人高10倍。本病患者易患肿瘤或白血病。

图9-3 Fanconi贫血的异常染色体
a示单位断裂;b示非同源染色体断裂后交换
3.毛细血管扩张性共济失调毛细血管扩张性共济失调(ataxiatelangiectasia,AT)为一种多见于儿童期的染色体隐性遗传病。1岁左右即可发病,表现为小脑性共济失调;6岁后眼和面、颈部出现瘤样小血管扩张。由于常有免疫缺陷,患者常死于感染性疾病。AT患者也有较多的染色体断裂。染色体畸变是非随机的,但常见有14/14易位或其它涉及14号染色体的改变。此外,B、D及G组的染色体重排也比较常见。患者的细胞对X线也特别敏感,其DNA修复能力明显下降。AT患者易患各种肿瘤,在45岁之前其患肿瘤的人数比正常人群增加3倍,主要是淋巴细胞白血病、淋巴瘤、网织细胞肉瘤等。人群中大约有1%AT的杂合子,他们占45岁以前死于肿瘤患者的5%。
4.着色性干皮病着色性干皮病(xerodermapigmentosum)患者皮肤对紫外线特别敏感,易出现多个的皮疹和色素沉着(图9-4)。患者染色体自发断裂率虽然没有增高。但在紫外线照射后明显上升,细胞也很容易死亡,存活下来的细胞由于DNA修复酶的缺陷而不能正常修复,常导致血管瘤、基底细胞癌等肿瘤发生。
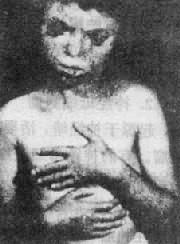
图9-4 着色性干皮病患者
五、肿瘤的遗传易感性
以上列举的许多事实都说明肿瘤发病中遗传因素的存在,因此肿瘤可以认为是基因染色体异常引起的疾病,其中一些遗传性肿瘤按照经典的孟德尔方式传递,但在更多情况下遗传的只是肿瘤的易感性,即易感基因,在个体易感染状态下如再发生体细胞突变,突变细胞就容易转化为肿瘤细胞。
个体的肿瘤遗传易感性是由特定的基因-染色体组合决定的。虽然对这些“易感基因(predisposing genes)”及其如何发挥作用了解得还不很清楚,但有一些事例表明它们可能通过生化的、免疫的和细胞分裂的机制促进肿瘤发生。
1.酶活性异常酶活性的改变可以影响致癌化合物在体内的代谢和灭活,例如芳烃羟化酶(arylhydrocarbon hydroxylase,AHH)它能在体内活化许多致癌的多环芳烃,包括从吸烟获得的各种芳烃,从而促进癌的发生。这种酶的可诱导性在人群中呈多态性,并按常染色体显性遗传(参阅第八章);另一方面酶的缺乏也可以导致对肿瘤的易感状态,例如着色性干皮病患者易患皮肤癌,这是由于DNA修复酶的缺陷导致细胞恶性变。
2.遗传性免疫缺陷机休正常的免疫监视(immuunesurveillance)系统不仅能抵御外来抗原的侵入,同时也能识别成为“异已”的突变细胞并加以排斥,免疫缺陷能使突变细胞得以逃脱这种监视而发展成为肿瘤。许多免疫缺陷患者都有易患肿瘤的倾向,例如无丙球蛋白血症(Bruton型)患者易患白血病和淋巴系统肿瘤等。
3.染色体病先天愚型患者急性白血病的发病率比正常人群高15-18倍。这两种疾病可能有共同的发病机制,即细胞分裂机制的紊乱。此外,Klinefelter综合征患者易患男性乳腺癌;一些两性畸形患者中(主要是表型女性而有XY核型者),由发育不全的性腺(睾丸的残留组织)也易发生精原细胞瘤和性母细胞瘤。
现今已知至少有200余种单基因决定的性状或疾病具有不同程度的易患肿瘤的倾向,这类疾病可称为遗传性癌前疾病。
第二节 肿瘤的染色体异常
很久以前已注意到,几乎所有肿瘤细胞都有染色体异常,且被认为是癌细胞的特征。自1960年在慢性粒细胞白血病(CML)患者发现了Ph染色体后,对肿瘤染色体异常的研究已发展为遗传学的一个分支,即肿瘤细胞遗传学。它的任务是阐明染色体畸变与肿瘤之间的关系,同时把获得的知识用于临床,如通过染色体检查来协助肿瘤的诊断、鉴别、预后和指导治疗。
一个肿瘤的瘤细胞染色体常有许多共同的异常,这可以用它们都来源于一个共同的突变细胞,即肿瘤发生单克隆学说来解释。但是癌细胞群体又受内外环境的影响而处于不变异之中,因此这些细胞的核型常常不完全相同,而且在同一肿瘤的发展过程中,核型也可以不演变。一些染色体畸变致死性的,而另一些畸变却能使细胞获得生长优势,因之肿瘤细胞群体经常处于选择和演变之中。肿瘤细胞群通过淘汰和生长优势,逐渐形成占主导地位的细胞群体,即干系(stem line)。干系的染色体数称为众数(modal number)。干系以外有时还有非主导细胞系,称为旁系(side line)。然而由于条件改变,旁系可以发展为干系。有的肿瘤没有明显的干系,有的则可以有两个或两个以上的干系。
1.肿瘤的染色体数目异常正常人体细胞为二倍体细胞,肿瘤细胞多数为非整倍体。非整倍体有两种情况:①染色体虽然不是46但在46上下,比46多的称超二倍体(hyperdiploid),比46少的称亚二倍体(hypodiploid)。瘤细胞染色体的增多或减少并不是随机的。例如许多肿瘤比较常见到的是8、9、12和21号染色体的增多或7、22、Y染色体的减少。②染色体数成倍地增加(3倍、4倍)称为高异倍性,但通常不是完整的倍数,故称为高异倍性(hyperaneuploid)(图9-5)。许多实体肿瘤染色体数或者在二倍体数上下,或在3-4倍数之间,而癌性胸腹水的染色体数变化更大。
肿瘤染色体异常的另一个特点是,即使在一个肿瘤中,各瘤细胞的染色体也不完全相同,甚至差别较大,但大多数肿瘤都可以见到1、2个干系,干系细胞的百分比并不固定。
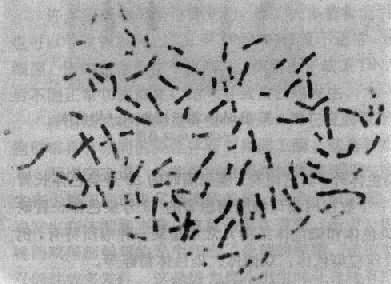
图9-5 一个癌细胞的染色体共104条,包括许多异常的染色体
2.肿瘤的染色体结构异常在56种人数肿瘤中发现3152种染色体结构异常,包括易位、缺失、重复、环状染色体和双着丝粒染色体等。结构异常的染色体又称为标记染色体(marker chromosome)。标记染色体分为2种:一种是非特异性的,它只见于少数肿瘤细胞,对整个肿瘤来说不具有代表性;另一种是特异性的,它经常出现在某一类肿瘤,对该肿瘤具有代表性。特异标记染色体的存在支持肿瘤起源于一个突变细胞的设想,现将最重要的特异标记染色体介绍如下:
(1)Ph染色体(费城1号染色体):Nowell及Hungerford于1960年发现慢性粒细胞性白血病(CML)血中有一个小于G组的染色体,由于首先在美国费城(Philadelphia)发现,故命名为Ph染色体。最初认为是22号染色体的长臂缺失所致,后经显带证明是9号和22号染色体长臂易位的结果。易位使9号染色体长臂(9q34)上的原癌基因abl和22号染色体(22q11)上的bcr(break point cluster region)基因重新组合成融合基因。后者具有增高了的酪氨酸激酶活性,这是慢性粒细胞性白血病的发病原因。Ph的重要临床意义在于:大约95%的慢性粒细胞性白血病病例都是Ph阳性,因此它可以作为诊断的依据,也可以用以区别临床上相似,但Ph为阴性的其它血液病(如骨髓纤维化等)。有时Ph先于临床症状出现,故又可用于早期诊断。此外,已知Ph阴性的慢性粒细胞性白血病患者对治疗反应差,预后不佳。
(2)14q+染色体:在90%的Burkitt淋巴瘤(非洲儿童恶性淋巴瘤)病例中可以见到一个长臂增长的14号染色体(14q+)。这是一条8号染色体长臂末端的一段(8q24)易位到了14号长臂末端(14q32),形成了8q-和14q+两个异常染色体(图9-7)。
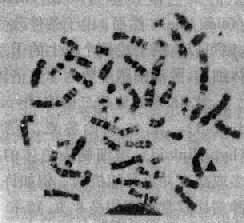
图9-6 Ph染色体示 9;22易位(9q34;22q11);→fi 22q-;▲示9+(晏炬提供)
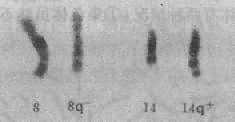
图9-7 Burkitt淋巴瘤的14g+染色体8q24;14q32易位
除了上述两个高度特异性标记染色体外,其它尚有:脑膜瘤时的22号染色体长臂缺失(22q-)或整个22丢失(-22);少数视网膜母细胞瘤患者的13号染色体长臂缺失(13q-等(表9-1)。另有一些标记染色体和染色体结构异常不是某一肿瘤所特有,例如巨大亚中着丝粒染色体、巨大近端着丝粒染色体、双微体、染色体粉碎等。
表9-1一些肿瘤常见的染色体异常
| 病名 | 染色体异常 |
| 慢性粒细胞白血病 Burkitt淋巴瘤 急性非淋巴细胞白血病 慢性淋巴细胞白血病 急性淋巴细胞白血病 恶性淋巴瘤 小细胞肺癌 卵巢乳头状腺癌 神经母细胞瘤 脑膜瘤 Wilms瘤 睾丸癌 畸胎瘤 | Ph,即t(9;22) t(8;14),t(2;8),t(8;22) +8;7q,5q或-5 t(8;21),t(15;17),t(9;22) t(11;14),+12 t(?;11),t(1;9),t(7;12),t(9;14) t(8;14),t(4;11),+21 t(4;11),+12 14q+,+12 del(3)(p14-23) t(6;14) del或t(1;?)(p 32-36;?) 13q -22,22q 11p 1(12p) 1(12p) |
3.脆性部位在人类染色体上还有一些易发生断裂的部位,称为可遗传的脆性部位(fragile sites)。其中一些与瘤细胞染色体异常的断裂点一致或相邻,另一些与已知癌基因的部位一致或相邻,它们与肿瘤的关系尚待阐明。
尽管人们很早就认识到染色体异常在肿瘤发生中可能起重要的作用,但只是在癌基因和肿瘤抑制基因发现后,其作用机制才逐渐明确。
第三节 肿瘤发病的遗传机理
一、体细胞突变
肿瘤可以看作是在个体遗传素质的基础上,尤其是在个体对肿瘤的遗传易感性基础上,致癌因子引起细胞遗传物质结构或功能异常的结果。这种异常大多数不是由生殖细胞遗传得来,而是在体细胞中新发生的基因突变所致。发生突变的癌前细胞在一些促癌因素的作用下发展为肿瘤。因此,有人认为,多数为肿瘤可看成是一种体细胞遗传病。
在自然界,基因突变是经常发生的,突变如果发生在与细胞增殖有关的基因,就可能导致细胞摆脱正常的生长控制,表现出恶性细胞的表型性状。
许多致癌物都是致突变物。它们大多数能引起DNA的损伤。这些损伤可以修复,也可以导致细胞死亡。如果DNA的修复不正常,细胞虽可继续存活,但却成了潜在的癌细胞,例如在着色性干皮病时,细胞由于缺乏DNA修复酶,因而在DNA被紫外线损伤后不能正常切除修复,结果导致皮肤癌发生。
肿瘤既然是体细胞突变的结果,则它就起源于单个突变细胞(单克隆)。许多肿瘤细胞群都具有相同的染色体畸变和同工酶,就是肿瘤发生的单克隆学说的证据。
两次突变说 一些细胞的恶性转化需要两次或两次以上的突变。第一次突变可能发生在生殖细胞或由父母遗传得来,为合子前突变,也可能发生在体细胞;第二次突变则均发生在体细胞本身,这就是两次突变(击中)说。两次突变说对一些遗传性肿瘤,如视网膜母细胞瘤的发生作出了合理解释。遗传型的视网膜母细胞瘤发病很早,并多双侧性或多发性。这是因为患儿出生时全身所有细胞已有一次基因突变,只需要在出生后某个视网膜母细胞再发后一次突变(第二次突变)。就会转变成为肿瘤细胞,故较易表现为双侧性或多发性。非遗传型视网膜母细胞瘤的发生则需要同一个细胞在出生后积累两次突变,而且两次都发生在同座位,因而概率很小,因此发病较晚,不具有遗传性,并多为单侧性,但该座位如果已发生过一次突变,则较易发生第二次突变,这也是非遗传型肿瘤不是太少的原因。
一些学者认为,体细胞癌变并不一定有基因结构的改变,当基因以外的物质或因素如蛋白质、RNA、生物膜发生了改变,而这些改变也能使与生长、分休有关的基因异常关闭或启动,此时,细胞也能转化为癌细胞,这一观点称为基因外调节学说。近年来对遗传印记的研究表明,基因的修饰如甲基化也能影响肿瘤有关基因的表达,从而促进细胞的转化。
二、癌基因
与肿瘤发生相关的基因可分为两大类:一类称为癌基因(oncogenes),它们能促进细胞的生长和增殖;另一类称肿瘤抑制基因(tumor-suppresor gene)或抑癌基因,它们能调节细胞的生长和分化而抑制肿瘤的发生。这两类基因的作用正好相反。它们的异常,或增强细胞生长和增殖,或去除正常的生长抑制,结果都会导致肿瘤发生。
癌基因:在致瘤病毒、人体和动物肿瘤都发现了能导致细胞恶性转化的核酸片段,称之为癌基因。来自病毒的称为病毒癌基因(v-onc),来自细胞的称为细胞癌基因(c-onc)或原癌基因(proto-oncogene),它们具有转化的潜能,可被激活成为癌基因。后来发现从酵母菌到人类的正常细胞几乎都有与之类似的片段。这种进化上的高度保守性表明它们具有重要的生物学意义。
最早发现的病毒癌基因是鸡Rous肉瘤病毒的v-src基因,以后又发现了与之同源的、见于正常鸡细胞的细胞癌基因(c-src)。许多癌基因都以最先见于的病毒来命名,如见于猴肉瘤病毒(Simian sarcoma)的称为sis基因,见于鸟类髓细胞增多症病毒(Myelocytomatosis)的称为myc基因等。最早发现的人类癌基因是H-ras基因,它是通过用人膀胱癌细胞系提取的DNA片段转染小鼠细胞,引起后者恶性转化而证实。以后在其他许多肿瘤和白血病也证明了人癌基因的存在。
1.癌基因的功能和分类已知的原癌基因已近100种,其中许多已定位不同的染色体区带。这些基因与细胞的生长、增殖等基本功能有关。它们或编码生长因子、生长因子受体和蛋白激酶而在生长信号的传递和细胞分裂中发挥作用;或者编码DNA结合蛋白而参与基因的表达或复制的调控(表9-2)。因此,可以按原癌基因的产物将其分为若干类型。如以src为代表的酪氨酸激酶类,以ras 为代表的G蛋白类,以myc为代表的核蛋白类,以sis为代表的生长因子类,以reb为代表的生长因子受体类等。原癌基因在个体发育或细胞分裂的一定阶段十分重要,但在成体或平时却不表达或表达受到严格的控制,当其发生突变或被异常激活时,产生的癌蛋白在性质或数量上异于正常,就可能导致细胞发生恶性转化。
表9-2 一些重要的癌基因
| 癌基因 | 来源 | 染色体定位 | 生化性质 |
| 信号传导蛋白 | |||
| abl | Abelson鼠白血病病毒 | 9q34 | 蛋白激酶 |
| src | 鸡Rous肉瘤病毒 | 20q12-13 | 蛋白激酶 |
| trk | 人大肠癌 | 1q32 | 蛋白激酶 |
| H-ras | Harvery大鼠肉瘤病毒 | 11p15.5 | GTPase |
| N-ras | 各种人类肿瘤(如神经母细胞瘤) | 1p13 | GTPase |
| 分泌生长因子 | |||
| sis | 猴肉瘤病毒 | 22q12.3-13.1 | 血小板生长因子(PLGF)β链 |
| 生长因子细胞 | |||
| 表面受体 | |||
| erb-A | 17q21-22 | 固醇受体 | |
| 核蛋白结合蛋白 | |||
| myc | 鸡肉瘤 | 8q24 | 结合DNA |
| N-myc | 人神经母细胞瘤 | 2p24 | 未知 |
2.癌基因的激活癌基因可以通过多种方式被激活而过度表达。
(1)突变激活:体细胞内的原癌基因可以因点突变而成为癌基因,产生异常的基因产物;也可由于点突变使基因摆脱正常的调控而过度表达。因此,突变激活又称为激活的质变模式(qualitative model)。例如在膀胱癌细胞株由于癌基因ras的12位密码子GGC变为GTC,使甘氨酸变为缬氨酸,结果导致细胞具有转化细胞的特征。现今已在膀胱癌、结肠癌等许多肿瘤发现了ras基因,后者编码一种膜蛋白,称为p21蛋白。
(2)易位激活:易位导致癌基因的重排或融合,产生异常的蛋白而使细胞转化。例如慢性粒细胞白血病9;22易位,形成了一种结构与功能异常的融合基因bcr-abl。它编码的蛋白能促成细胞的恶性转化。
原癌基因通过易位插到强力的启动子附近也可导致激活。Burkitt淋巴瘤的t(8;14)即c-myc癌基因由8号染色体易位到14号染色体的免疫球蛋白重链基因附近,易位使c-myc置于免疫球蛋白H链基因的启动子控制下。免疫球蛋白基因是一个活跃的基因,为了抵抗进入体内的各种抗原,它不断编码各种抗体蛋白,其启动子特别活跃,因而易位的c-myc基因转录活性明显增高。增多的myc蛋白质使一些控制生长的基因活化,最终导致细胞恶变。
(3)癌基因扩增:原癌基因还可因某种原因自身扩增而过度表达。在肿瘤细胞尤其是胚胎神经组织肿瘤细胞中有时见到的双微体(double minutes)和染色体上的均染区 (homogenouslystaining region)就是原癌基因DNA片段扩增的表现。例如,在肿瘤细胞中c-myc癌基因可扩增数百到数千倍。
三、肿瘤抑制基因
肿瘤抑制基因又称为抑癌基因或抗癌基因(anti-oncogenes)。它们的功能是抑制细胞的生长和促进细胞的分化。当两个等位都因突变或缺失而丧失功能,即处于纯合失活状态时,细胞就会因正常抑制的解除而恶性转化。视网膜母细胞瘤的Rb基因就是一个典型的例子。遗传型视网膜母细胞瘤患者出生时Rb基因的一个等位基因由于生殖细胞突变而丧失功能,出生后如视网膜母细胞中另一个等位基因发生了体细胞突变,这个细胞就会转化为肿瘤细胞。现今在不少的肿瘤均已证实了相应的抑制基因的存在、缺失及其在肿瘤发病中的作用(表9-3)。这些肿瘤在临床上都呈染色体显性遗传方式,但从基因水平上看,它们都是隐性的,因两个等位基因均有突变才能使细胞恶性转化。
视网膜母细胞瘤(Rb)基因定位于13号染色体1区4带,全长约200kb,有27个外显子,编码924个氨基酸的核磷蛋白(Rb蛋白),分子量为110kb,具有抑制肿瘤细胞增殖生长的作用。Rb基因的缺失或功能丧失不仅见于视网膜母细胞瘤,而且还见于骨肉瘤、小细胞肺癌、乳腺癌等肿瘤中。
表9-3 肿瘤抑制基因及其产物
| 肿瘤抑制基因 | 染色体定位 | 基因产物及其功能 | 基因异常引起的肿瘤 | |
| 遗传型 | 散发型 | |||
| Rb1 | 13q14 | p110细胞周期调节 | 视网膜母细胞瘤 | 小细胞肺癌等 |
| WT1 | 11p13 | 锌脂蛋白,结合DNA | Willm瘤 | 肺癌等 |
| p53 | 17p13.1 | p53,细胞周期调节 | Li-Frameni综合征 | 肺癌,乳腺癌等 |
| NF1 | 17p11.2 | GTP酶活化蛋白 | 神经纤维病,Ⅰ型 | - |
| DCC | 18q21 | 参与细胞表面作用 | 未知 | 结肠直癌 |
| APC | 5q21 | 家族性腺瘤息肉 | ||
| MCC | 5q21-22 | 家族性腺瘤息肉 | ||
如果抑癌基因的纯合型丢失是基因缺失的结果,可以通过比较正常组织与癌组织有关DNA片段的杂合性丢失(loss of heterozygosity,LOH)来证实。例如,正常组织的DNA等位片段与特定内切酶和特定的探针杂交后证明是杂合的,而癌组织的等位片段经证明是纯合的,表明抑癌基因发生了杂合丢失。
然而并非所有的抑癌基因都必需有两个等位基因的丧失才导致肿瘤发生。现知APC基因在这一过程中作用。同图可见,在肿瘤发生发展的过程中既有肿瘤抑制基因的丢失,也有癌基因的活化,而且不止一种。如结肠癌发生的过程中,就有多个基因的异常,其中包括ras癌基因和p53、APC和MCC等几个抑癌基因。这些改变中任何一种基因异常如果是遗传的,这就构成了肿瘤的家族性易感性的基础。
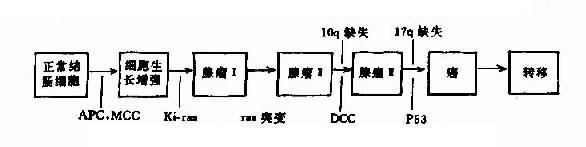
图9-8 结肠癌发生的各个阶段和肿瘤相关基因的异常
恶性肿瘤的转移是一个复杂的过程,包括瘤细胞由原发肿瘤脱落,进入细胞外基质和血管或淋巴管,并在远处适宜的组织中生长。近年来的研究发现,存在着促进转移的转移基因(metastatic genes)和抑制转移的基因(metastasissuppressor genes)。
1.转移基因一些编码细胞表面受体的基因可能和瘤细胞的转移有关。例如整合素(integrins)是一类细胞表面粘合受体,能识别细胞基质中的粘附蛋白,起着固定细胞抑制其迁移的作用。可以设想,这些受体基因的突变和失去功能将有利于瘤细胞的转移。
瘤细胞的浸润能力与其分泌的能降解基质的蛋白有关,已知内糖苷酶和Ⅳ型胶原酶能降解基底膜中的相应成分,增加瘤细胞侵袭基底膜的能力。
最后,用许多癌基因ras、fos、mos、src,或突变了的肿瘤抑制基因如p53基因的片段转染培养中的细胞,都可提高这些细胞的浸润和转移能力。
2.转移抑制基因一些基因编码的蛋白酶能够直接或间接地抑制具有促进转移的蛋白。例如金属蛋白组织抑制因子基因(TIMP)编码一种糖蛋白,能与转移密切有关的胶原酶结合,降低瘤细胞的侵袭和转移能力。
在人和小鼠中已发现nm23基因的表达与乳腺癌等肿瘤的转移密切相关。nm23基因编码一个17kd的蛋白质,基因在转移的乳腺癌中表达高,而在无转移的肿瘤中表达低,表明nm23是一个肿瘤转移抑制基因。至于它如何抑制肿瘤转移还有待阐明。
除了肿瘤细胞外,受体细胞的一些基因在转移灶生成中也具有一定意义。由此可见转移基因、转移抑制基因与宿主有关基因在表达最终决定于肿瘤细胞的转移。
综上所述,肿瘤的发生发展是一个复杂的生物学过程,它是细胞遗传物质异常的结果,同时也涉及机体的内环境中的各种因素,包括机体的免疫能力、各种生长因子和生物活性物质,而它们都是基因表达的结果,其中癌基因与抑癌基因的异常起着关键的作用。
第十章 遗传病的诊断
遗传病的诊断(diagnosis of hereditary disease)可分为产前诊断、症状前诊断和现症病人诊断三种类型。遗传病的确诊是开展遗传咨询和防治工作的基础。遗传病诊断方法有普遍性诊断原则,又有遗传学的特殊诊断手段。普遍性诊断原则是与诊断一般疾病相同的方法,即通过对病史、症状、体征、实验室检查和其他诊断技术所获得的资料进行归纳分析,同时排除拟诊疾病,然后确立诊断。但由于遗传病种类繁多,特殊的病因分子基础,以及不同的遗传病存在许多相似的症状和体征,故除一般诊断方法外,尚需辅以遗传学的特殊诊断手段,如染色体和性染色体检查、肤纹分析、携带者的检出等。此外,近年来发展起来的产前诊断技术对遗传病诊断、遗传咨询和防治工作都极为重要。
第一节 病史、症状和体征
1.病史由于遗传病多有家族聚集现象,所以病史采集的准确性关系重要。除一般病史外,应着重患者的家族史、婚姻史和生育史。
(1)家族史:采集家族史时应特别注意因患者和代诉人由于文化程度、记忆能力、思维能力、判断能力及精神状态而使症状、体征的描述不够准确或不全面,或因患者或代诉人提供假材料等都会影响家族史材料的准确性。
(2)婚姻史:着重了解婚龄、次数、配偶健康情况以及是否近亲结婚。
(3)生育史:着重询问生育年龄、子女数目及健康状况,有无流产、死产和早产史,如有新生儿死亡或患儿,则除询问父母及家庭成员上述情况外,还应了解患儿有无产伤、窒息,妊娠早期有无患病毒性疾病和接触过致畸因素,如服过致畸药或接触过电离辐射或化学物质史。
2.症状和体征遗传病有其它疾病相同的体征和症状,往往又有其本身特异性症候群,为诊断提供初步线索。如患有智力发育不全伴有特殊腐臭尿液提示苯酮尿症;智力发育不全伴有白内障,肝硬化等提示乳糖血症;智力低下,伴有眼距宽、眼裂小、外眼角上斜等体征要考虑先天愚型;智力发育不全伴有生长发育迟缓、五官、四肢、内脏等方面的畸形提示可能为常染色体病;若有性腺发育不全或有生殖力下降、继发性闭经、行为异常的可疑为性染色体病。
由于大多数遗传病在婴儿或儿童期即可有体征和症状表现,故除观察外貌特征外,还应注意身体发育快慢、体重增长速度、智力增进情况、性器官及第二性征发育状态、肌张力强弱以及啼哭声是否异常等。
应当指出,由于遗传普通存在遗传异质性,若欲作出病因诊断,单凭症状和体征资料是很困难的,因此必须加强实验室检验和辅助器械检查才能提高遗传病的诊断水平。
第二节 系谱分析
在遗传病诊断时进行系谱分析有助于区分单基因病和多基因病,以及属于哪一种遗传方式;有助于区分某些表型相似的遗传病以及由于遗传性而出现的不同遗传方式。进行系谱分析应注意下列问题:①系谱的系统性、完整性和可靠性。系谱分析时必须有一个系统完整和可靠的系谱,否则可以导致错误的结论。完整的系谱应有三代以上有关患者及家庭的情况。有关成员要逐个查询,特别是关键不可遗漏,死亡者(包括婴儿死亡)须查清死因,是否近亲婚配、有无死胎、流产史,并记录在系谱中。在家系调查过程中避免由于患儿或代诉人不合作或提供假情况,例如不愿提供重婚、非婚子女、同父异母、同母异父、养子养女等,以致错给系谱,必要时应对患者亲属进行实验室检查和其他辅助检查使诊断更加可靠。②分析显性遗传病时,应注意对已知有延迟显性的年轻患者,由于外显不全而呈现隔代遗传现象进,不可误认为是隐性遗传。③新的基因突变。有些遗传家系中除先证者外,家庭成员中找不到其他的患者,因而很困难从系谱中判断其遗传方式,更不可因患者在家系中是“散发的”而定为常染色体隐性遗传。如假肥大型肌营养不良是一种致死的X连锁隐性遗传病,约有1/3的病例为新的基因突变引起。④显性与隐性概念的相对性。同一遗传病可采用的观察指标不同而得出不同的遗传方式,从而导致发病风险的错误估计。如镰形细胞贫血症在临床水平,纯合子(HbSHbs)有严重的贫血,而杂合子(HbAHbs)在正常情况下无贫血,因此,这时突变基因(HbS)对HbA来说被认为是隐性的;然而,当杂合子的红细胞处于氧分压低的情况下,红细胞亦可形成镰刀状,所以在细胞数目水平,观察红细胞呈现镰刀状,此时Hbs对HbA来说是显性的。但从镰形细胞数目理解,来自杂合子的红细胞形成少量镰形细胞,其数目介于正常纯合子(HbAHbA)与突变基因纯合子(HbSHbs)之间故呈不完全显性遗传。遗传方式不同,对后代复发风险估计也应不同。
此外,在系谱分析统计子女发病比值时应校正因统计带来的偏倚。
第三节 细胞遗传学检查
1.染色体检查染色体检查亦称核型分析(karyotypeanalysis)是确诊染色体病的主要方法。目前随着显带技术的应用以及高分辩率染色体显带技术的出现和改进,能更准确地判断和发现更多的染色体数目和结构异常综合征,还可以发现新的微畸变综合征。值得注意的是,染色体检查应结合临床表现进行分析才能得出正确诊断。
染色体检查标本的来源,主要取自外周血、绒毛、羊水中胎儿脱落细胞和脐血、皮肤等各种组织。
染色体检查的指征:①有明显的智力发育不全、生长迟缓或伴有其他先天畸形者;②夫妇之一有染色体异常,如平衡结构重排、嵌合体等;③家族中已有染色体或先天畸形的个体;④多发性流产妇女及其丈夫;⑤原发性闭经和女性不育症;⑥无精子症男子和男性不育症;⑦两性内外生殖畸形者;⑧疑为先天愚型的患儿及其父母;⑨原因不明的智力低下伴有大耳、大睾丸和(或)多动症者;⑩35岁以上的高龄孕妇(产前诊断)。
2.性染色质检查优点是方法简单,主要用于疑为两性畸形或性染色体数目异常的疾病诊断或产前诊断,有一定价值,但确认仍需依靠染色体检查。
性染色体检查材料来自发根鞘细胞,皮肤或口腔上皮细胞,女性阴道的上皮细胞,也可取自绒毛和羊水的胎儿脱落细胞涂片等。
X染色质数目计数分析,可适用于X染色体异常而引起的性染色体畸形综合征的检出(参阅第二章)。Y染色质数目计数分析可适用于具有一个或一个以上Y染色体的个体。如正常男子只有1个Y染色质,而XYY男性有2个,XXYYY男性有3个;真两性畸形(46,XX/46,XY)患者则X和Y染色质均为阳性(各1个)。
第四节 基因及基因产物分析
基因突变引起的单基因病往往表现在酶和蛋白质的质和量的改变或缺如。因此,酶和蛋白质的定性定量分析是诊断单基因病或分子代谢病的主要方法。根据分子代谢病的临床特点可以从三个层次检测和分析。
1.代谢产物的检测酶缺陷导致一系列生化代谢紊乱,从而使代谢中间产物、底物、终产物旁路代谢产物发生变化。因此,检测某些代谢产物的质和量的改变。可间接反映酶的变化而作出诊断。例如疑为苯酮尿症患者,可检测血清苯丙氨酸或尿中苯乙酸浓度;粘多糖病可测定尿中硫酸皮肤素、硫酸乙酰肝素;DMD可检测血清中磷酸肌酸激酶活性作出诊断等。
2.酶和蛋白质的分析基因突变引起的单基因病主要是特定的酶和蛋白质的质和量改变的结果。因此,对酶活性的改变和蛋白质含量测定是确诊某些单基因病的主要方法。随着生化检测技术的不断进步,还可对酶和蛋白质的结构变异型(variants)作出鉴定。
检测酶和蛋白质的材料主要来源于血液和特定的组织、细胞,如肝细胞、皮肤成纤维细胞、肾、肠粘膜细胞等。但应注意,一种酶缺乏不一定在所有组织中都能检出,例如苯丙氨酸羟化酶必须用肝活检,而在血细胞中无法得到。
3.基因分析基因诊断(gene diagnosis)是利用DNA分析技术直接从基因水平(DNA或RNA)检测遗传的基因缺陷。它和传统的诊断方法主要差异在于直接从基因型推断表型,即可以越过产物(酶和蛋白质)直接检测基因结构而作出诊断,这样就改变了传统的表型诊断方式,故基因诊断又称为逆向诊断(reverse diagnosis)。这一诊断方法不仅可对患者,还可以在发病前作出症状前基因诊断,也可对有遗传病风险的胎儿作出生前基因诊断。此外基因诊断不受基因表达的时空限制,也不受取材的细胞类型和发病年龄的限制。这一技术还可以从基因水平了解遗传病异质性,有效地检出携带者。因此近年来这一技术日新月异地迅速发展,并已经在遗传病诊断中发挥了巨大作用(参阅第十三章)。
第五节 产前诊断
产前诊断(prenatal orantenatal diagnosis)又称宫内诊断(intrauterine diagnosis)是对胚胎或胎儿在出生前是否患有某种遗传病或先天畸形作了准确的诊断。产前诊断的适应症的选择原则,一是有高风险而危害较大的遗传病;二是目前已有对该病进行产前诊断的手段。几种主要产前诊断方法如下:
1.X线检查主要用于检查18周以柏胎儿骨骼先天畸形。但因X线对胎儿有一定影响,现已极少使用。
2.超声波检查是一项简便对母体无痛无损伤的产前诊断方法。B型超声波应用最广,利用超声波能作出的产前诊断或排除性诊断见表10-1。此外还可直接对胎心和胎动进行动态观察,并可摄象记录分析,亦可作胎盘定位,选择羊膜穿刺部位,可引导胎儿镜操作,采集绒毛和脐带血标本供实验室检查。
3.胎儿镜胎儿镜(fetoscope)又称羊膜腔镜或宫腔镜,是一种带有羊膜穿刺的双套管光导纤维内窥镜。能直接观察胎儿,可于怀孕15-21周进行操作。主要用于胎儿血的取样、活检和产前诊断,利用皮肤活检可诊断8种以上的遗传性皮肤病,也可对胎儿形态异常进行观察。此外,胎儿镜还可判定胎儿性别和对某些遗传病进行宫内治疗。由于B超的应用,此方法已少用。
4.羊膜穿刺术羊膜穿刺(amniocentesis)亦称羊水取样。抽取羊水最佳时间是妊娠16-20周。历为此时羊水量多、胎儿浮动,穿刺时进针容易,且不易伤及胎儿。羊水中有胎儿脱落细胞,经体外培养后,可进行染色体分析、酶和蛋白质检测、性染色质检查提取DNA作基因分析,也可不经培养,用微量技术作酶和蛋白质分析或直接提取DNA作基因诊断。
5.绒毛吸取绒毛可经宫颈部取样,最好在B超监视下进行。绒毛取样(chorionic villi aspiration sampling,cvs)(图10-1)一般于妊娠9-11周时进行。绒毛枝经处理(与蜕膜严格分离)或经短期培养后进行染色体分析、酶和蛋白质检测和直接抽取DNA进行基因分析。
表10-1 或利用超声波检查作产前诊断的疾病
| 1.水肿 水肿胎 羊水过多或羊水过少 2.脸和颈 肋裂囊 腭裂、唇裂 水囊状淋巴管瘤 眼距宽 小颌 3.中枢神经系统 无脑畸形 脑膨出 全前脑无裂症 积水性无脑畸形 脑积水 脊髓膜膨出 小头畸形 4.胸 先天性心脏病 肺腺囊肿样畸形 膈疝 胸膜积液 小胸腔 5.腹 十二指肠闭锁 | 食管闭锁 腹裂畸形 脐膨出 6.肾 多囊肾 肾发育不全 肾盂积水 7.骨骼异常 无指(趾)畸形 缺指(趾) 多指骨折 缺肢畸形 8.骨骼发育不良 软发育不良 胸部发育不全窒息 前肢曲骨发育不良 磷酸酶过少症(幼儿型) 脊柱后凸 成骨不全 短肋多指型(Saldino-Noonan) 短肋多指型(Majewski) 先天性脊柱骨髓发育不良 致死性发育不良 遗传性血小板减少症伴桡伴骨缺失 |
图10-1 羊膜穿刺抽取羊水示注射器穿越腹壁到羊腔抽取羊水
6.脐带穿刺术(cordocentesis)经母腹抽取胎儿静脉血,可在B超引导下于孕中期、孕晚期(17-32周)进行。这项技术在我国已远较国外普及,成功率高,也较安全。脐血可作染色体或血液学各种检查,亦可用于因羊水细胞培养失败,DNA分析无法诊断而能用胎儿血浆或血细胞进行生化检测的疾病,或在错过绒毛和羊水取样时机下进行,在一些情况下,可代替基因分析。例如,α地贫可直接测定Hb Barts;血友病可直接测定凝血因子Ⅷ.
图10-2 绒毛吸取术示借助超扫描器用能屈的套管或腰椎穿刺针吸取绒毛
7.孕妇外周血分离胎儿细胞这是一项非创伤性产前诊断技术,并易于被孕妇接受。孕妇外周血中的胎儿细胞至少有3种,即滋养叶细胞、有核红细胞和淋巴细胞。目前许多学者都致力于解决胎儿细胞的识别、富集和如何排除母血的“污染”等。在孕妇外周血中的胎儿细胞数量虽然不多,但已有用单克隆抗体或以滋养叶细胞表面特异性抗原的抗体作为标记等来识别胎儿细胞。结合富集和纯化技术不断完善,如果方法能更加简便、经济,将可能在遗传病的预防中发挥作用。
8.植入前诊断植入前诊断(preimplantationdiagnosis)是利用微操作技术和DNA扩增技术对胚泡植入前进行检测。获得植入前胚胎的主要方法是:子宫冲洗和体外授精。植入前诊断的基本技术包括:①卵裂球的微活检:即从2-8个细胞期的胚胎细胞中分离出单个细胞进行检测;②胚胎的冻存:如果微活检技术快速,亦勿需冻存即可送回子宫;③卵裂球的培养:其目的在于得到更多的细胞,有利于诊断。目前已有用酶超微量分析测定HGPRT诊断Leach-Nyhan综合征;用PCR技术(参阅第十三章)作镰形细胞贫血、甲型血友病、性别检定、DMD、β-地中海贫血等单基因的产前诊断,虽然目前仅有个别成功先例,操作难度大,还不能用于临床,但前景是诱人的。
产前诊断中的实验室检查
1.细胞遗传学的产前诊断
(1)羊水细胞染色体检查:羊水细胞主要来自胎儿的皮肤、胃肠道、呼吸道、泌尿生殖道的粘膜脱落细胞。将羊水中的细胞经过常规培养后制备显带染色体以进行核型分析或脆性X染色体分析,而作出诊断。
(2)孕早期绒毛细胞染色体检查:直接法制备绒毛染色体虽简易可行,但可能有假阳性或假阴性,故其结果只作为初步筛查,应以培养法制备的结果为最终报告。
应用绒毛或羊水细胞可同时检查性染色质,以初步预测胎儿性别、性染色体数目异常等。
研究表明,羊水细胞核型分析为21三体者,其羊水及母血清的甲胎蛋白(AFP),母血雌三醇(E3)值低于正常平均值,而绒毛促性腺激素(HCG)值高于正常,因此有人建议除母龄>35岁外,检测母血AFP、E3和HCG三项联合数据从而定出高危先天愚型儿,再进行羊水细胞核型分析更好。
2.羊水的生化学检查羊水上清液进行生化学检查较难获得胎儿患某种遗传病的证据,但对了解胎肝、胎肾、胎肺成熟度有所帮助。甲胎蛋白和乙酰胆碱酯酶测定对检出患神经管缺陷的胎儿有益。对某些遗传代谢病高风险的孕妇测定羊水培养细胞有关本科的活性或可获得胎儿是否患病的信息。
3.神经管缺陷的产前诊断神经管缺陷(neuraltube defects,NTD)是一类胚胎发育过程中神经管封闭障碍或已封闭的神经管再穿孔所致的先天性畸形。除可通过B超进行产前诊断外,还可用下列方法检出:
(1)羊水甲胎蛋白(α-fetoprotein,AFP)测定:当胎儿为开放性神经管畸形时(如无脑儿、脊柱裂等),脑脊液中AFP可以直接进入羊水,使羊水AFP值升高,达10倍以上,可作诊断指标,诊断率达90%。但应注意,胎儿如因食管闭锁而不能吞咽羊水将AFP消化,或先天性肾病及脐疝等都可使羊水中AFP值升高。
(2)羊水乙酰但碱酯测定:乙酰胆碱酶(acetylcholine esterase,AchE)能特异性水解乙酰胆碱。胎儿期神经细胞分化未成熟,可溶性AchE进入胎儿脑脊液比成人多,当胎儿有开放性神经管缺陷时,胎儿脑脊液羊水间的通透增强,使羊水中AchE显著升高,测定AchE活性比测定AFP更为敏感可靠。
(3)超声波诊断:超声波扫描诊断开放性神经管缺陷,妇女无痛苦。指征:有生育过神经管畸形胎儿的孕妇;孕妇血清AFP初筛可疑者。由于B超诊断的普及,B超声有取代生化检查之势。
4.分子代谢病的产前诊断
(1)胎儿性别鉴定:如果孕妇为X连锁隐性遗传病(例如甲型血友病、DMD等)携带者,生育患子的概率5%。因此在没有条件进行基因诊断的情况下,可考虑对男胎进行选择性流产,防止患儿出生。可以采用羊水或绒毛细胞作染色体核型分析,或抽取细胞DNA用Y特异性引物PCR扩增(参阅第十三章)分析,或用B鉴定性别。扩增Y特异性片段,以判定胎儿性别,对防止X连锁遗传病患儿出生有一定意义。
(2)胎血、羊水、羊水细胞和绒毛细胞生化检测:见第四节。
(3)基因分析:由于某些酶不能在羊水绒毛细胞表达,或表达水平很低,这就必须抽取羊水细胞或绒毛细胞DNA进行基因诊断。随着研究工作迅速开展将会有更多的遗传病前基因诊断得以确诊(参阅第十三章)。
目前已知有100余种分子代谢病能通过绒毛组织或羊水细胞作出产前诊断。随着基因分析方法的不断简化和完善,分子代谢病的产前诊断正在由蛋白质和酶的检测过渡到分析某编码基因的突变,但检出很多等位基因突变时基因分析就越来越困难,而生化分析能检测源于任何等位基因突变所致蛋白质功能的异常,这种优点特别是对于高度异质性或新突变比例高的疾病显得特别突出。
第六节 皮肤纹理分析
1.人类正常的肤纹人体的皮肤由表皮和真皮组成。真皮乳头向表皮突起,构成许多整齐的乳头线称为嵴线(ridge),嵴线之间凹陷部分为沟(furrow)。指(趾)掌(脚)部位的皮肤表层因皮嵴和皮沟走向不同而形成各种皮肤纹理特征。所谓皮肤纹理(dermatoglyphy)亦称皮纹,即指人体皮肤某些特定部位出的纹理图形。
人体的皮肤纹理属多基因遗传,具个体的特异型。皮肤纹理于胚胎14周形成,一旦形成终生不变,所以皮纹具有高度稳定性特点。
(1)指纹类型(finger tip patterns):指纹是指手指端的纹理,依指端外侧三叉的有无数目分三种类型(图10-3)。
1)弓形纹(arch,A):由平等的弓形嵴纹从一侧走向另一侧,中间隆起弓形,无三叉隆起似帐篷状者,称为帐弓纹(tented arch,At)。
2)箕形纹(loop,L)嵴纹从一侧发出后向上弯曲,又转回发生的一侧,形似簸箕状。若萁口朝向手的尺侧称为尺箕(ulner loop,Lu)或称为箕,箕口朝向手的桡侧称为桡箕(radialloop,Lr)或反箕。箕头的侧下方有一个三叉。所谓三叉(triradius)是指肤纹中有三组不同走向的嵴纹汇聚在一处呈Y或人字形者。
3)斗形纹(whorl,W):特点是有两个或两个以上三叉,嵴纹走向是同心环形(环形纹)或走向同一侧(偏形纹),此类斗形纹称为双箕斗(dorble loop whorl,Wd)。上述其它非双箕斗为一般斗形纹(simplewhorl,Ws)。
(2)总指嵴纹数:从箕形纹或斗形纹的中心点到三叉画一直线,计数这条直线跨过的嵴纹数目,称为嵴纹计数(ridge count)。弓形纹无三叉,其嵴纹数为0,箕形纹有一个三叉故有一个嵴纹数,斗形纹有两个三叉故有二个嵴纹数(取两个中较大的为准),将十指嵴纹数相加,即为总指嵴纹数(total finger ridge count,TFRC)。






图10-3 各种指纹类型及嵴纹计数计数沿着中心点到三叉的直线
(3)掌纹:手掌中的皮纹称为掌纹(palmar print),比较重要的是轴三叉和∠atd的测定。
轴三叉(axialtriradius)是指在腕横褶线远侧,大致在环指直下,有一呈人字形的叉d,引一直线连接于轴三叉t所形成的夹角即∠atd。其大小表明t的具体位置(图10-4)。我国正常人的∠atd平均为41°。t的位置移近掌心,则atd角增大。∠atd56°以t″表示。
(4)褶线:褶线是指手指和手掌的关节弯曲活动处明显可见的褶纹,分别称为指褶线和掌褶线。它们虽不属皮肤纹理,但其变化在某些遗传病诊断中有一定价值。
图10-4 轴三叉及atd角的测量法
1)掌褶线(palmal flexion crease):正常人的手掌褶线有三条;远侧横褶线、近侧横褶线和大鱼际纵褶线。有时远侧横褶线和近侧横褶线连接成一条单一的褶线横贯全掌,称为猿线(simian crease),我国称为通贯手。然而有时相接的程度不同,可分为各种变异型(图10-5)。在我国正常人群中犯规线发生率可达3.53%-4.87%.
2)指褶线(digital flexion crease):正常人除拇指只有一条指褶线外,其余各指都有二条褶线。





图10-5 掌褶线的各种变异型
(5)拇趾球部纹型:人的脚趾和脚掌上的肤纹,称为趾纹和跖纺,但具有临床意义的只涉及拇趾球部(hallucal area)纹型。拇趾球部的肤纹图形弓形也有弓、箕、斗等各种图形,并按照肤纹的走向不同可分为下列主要类型:近侧弓、腓侧弓、胫侧弓、远侧箕、腓侧箕、胫侧箕及斗形纹,引外还有复合纹。
2.皮纹检查的临床意义皮纹变化与某些染色体异常、先天性疾病以及不明原因的综合征有一定相关,但它的变化不是特异的,故只能作为诊断旁证或疾病的初筛,以便进一步确诊。现叙述几种常见疾病的肤纹变化。


图10-6 拇指球部纹型
1.染色体病
(1)21三体综合征(先天愚型):患者手指头形纹频率减少,而箕形纹增多,特别是尺箕比例高,TFRC较少,小指常是单一指褶线,大约有一半患者出现通贯手,∠atd1/2),她是携带者的可能性减小(